
Abstract
The novel coronavirus disease that arises in the end of 2019 (COVID-19) in Wuhan, China, has rapidly spread over the globe and was considered as a world pandemic. Currently, various antiviral therapies or vaccines are available, and many researches are ongoing for further treatments. Targeting the coronavirus’ main protease (key enzyme: 3CLpro) is growing in importance in anti-SARS-CoV-2 drug discovery process. The present study aims at predicting the antiviral activity of two novel compounds using in silico approaches that might become potential leads against SARS-CoV-2. The 3D structures of the new compounds are elucidated by single-crystal X-ray techniques. The interactions between different units of 4 and 5 were emphasized by analyzing their corresponding Hirshfeld surfaces and ESP plots. NBO and FMO analyses were investigated as well. Molecular docking combined with molecular dynamics simulations (MDs) was performed to investigate the binding modes and molecular interactions of 4 and 5 with the amino acids of coronavirus main protease (6LU7) protein. The best docking scores were obtained for both ligands through the major binding interactions via hydrogen/hydrophobic bonds with the key amino acids in the active site: HIS41, CYS145, MET49, MET165, HIS172, and GLU166 amino acids. A MD simulation study was also performed for 100 ns to validate the stability behavior of the main protease 3CLpro-ligand complexes. The MD simulation study successfully confirmed the stability of the ligands in the binding site as potent anti-SARS-CoV-2 (COVID-19) inhibitors. Additionally, MMPBSA energy of both docked complexes was determined as a validation assay of docking and MD simulations to validate compound conformation and interaction stability with 3CLpro. The synthesized compounds might be helpful in the fight against COVID-19 prior to biological activity confirmation in vitro and in vivo.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a virus responsible for the present coronavirus disease 2019 (COVID-19) pandemic, has spread all across the globe since first reported in Wuhan, Hubei province of China. The virus was closely related to SARS-CoV in terms of genome homology and stands at seventh number in the family of Coronaviridae known to cause infection in humans [1]. By now, the virus has been reported from 219 countries and tested positive in over 103,045,690 people with 2,225,836 deaths [2, 3]. The primary route of transmission of the virus is small droplets expelled by affected person upon coughing, breathing, sneezing, and talking [2, 4]. Infected person either remains asymptomatic or develops typical COVID-19 symptoms of fatigue, fever, cough leading to difficulty in breathing, and loss of smell [5]. In severe cases, the mentioned symptoms progress to pneumonia, acute respiratory distress syndrome, and failure of multiple organs that results into death [6]. The death rate of the COVID-19 has been estimated in between 3 and 6%.
Coronaviruses are a member of Coronaviridae family, subfamily of Orthocoronavirinae in the order of Nidovirales. These viruses possess the largest RNA genome of 22–32 kb in length that is positive sense and single stranded [7, 8]. The RNA genome forms complexes with the nucleocapsid (N) protein to make a helical capsid. The SARS-CoV-2 has total of 11 genes which encode ORF2 (Spike protein), ORF4 (Envelope protein), ORF5 (Membrane protein), ORF9 (Nucleocapsid protein), ORF1ab, ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10 [9, 10]. Coronaviruses are known to cause mild to moderate upper respiratory tract and gastrointestinal illnesses in mammals and birds. Prior to SARS-CoV-2, six species (HCoV-NL63, HCoV-HKU1, HCoV-OC43, HCoV-229E, SARS-CoV, and Middle East respiratory virus coronavirus (MERS-CoV)) of this family had been previously known to cause human infections. Exception to SARS-CoV and MERS-CoV which have the potential to cause serious respiratory epidemics, the rest of species are endemic strains and associated with common cold [11,12,13]. SARS-CoV was the first reported coronavirus specie during first large-scale outbreak in 2003 and liable for 8098 confirmed cases and 774 deaths across 17 countries with fatality rate of 9.6% [14,15,16]. MERS-CoV is a zoonotic respiratory infection, first diagnosed in the Middle East in 2012 with reported 2494 infection cases and 858 deaths. As coronaviruses are highly prevalent, owing to their genetic multiformity, and high rate of recombination, these viruses poses a constant threat to humans [7, 17, 18]. While the epidemic linked to the SARS-CoV2 coronavirus has spread around the world, research is mobilized to accelerate the production of knowledge on this virus, on the disease it causes (Covid-19) as well as the means of cure it and prevent it.
Since the beginning of COVID-19 crises, several different classes of drugs are reported against SARS-COV-2. For example, using drug repurposing approach, Skariyachan et al., (2020) reported FDA-approved drugs, chloroquine, hydroxychloroquine, favipiravir, lopinavir, remdesivir, and ritonavir, against 15 druggable proteins of SARS-CoV-2. They reported that ritonavir and lopinavir are better binders of the SARS-CoV-2 targets based on in silico molecular docking, molecular dynamics (MD) simulation, and binding free energy methods [19]. In another study, solanine, acetoside, and rutin natural compounds were reported as dual inhibitors of SARS-CoV-2 main protease and Spike proteins [20]. Remdesivir is revealed to show strong binding with the main protease enzyme and suggested to act as a template for novel furin protease inhibitors [21]. Using comprehensive in silico approaches, a virtually identified compound, ABBV-744, was reported as a potential inhibitor of SARS-CoV-2 main protease [22].
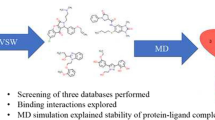
In the present study, heterocyclic derivatives are synthesized (4 and 5) and are evaluated as potential functional blockers of SARS-CoV-2 3C-like protease (3CL-pro) enzyme using an integrated approaches of X-ray crystallography, DFT calculation, molecular docking study, and MD simulation approaches. The molecular structures of 4 and 5 were elucidated through X-ray crystallography. Also, the Hirschfeld surface and electrostatic potential surfaces (EPS) of molecules 4 and 5 were reported in order to identify the key regions of molecules contributing to the interactions with 3CLpro protein. On the other hand, the molecular docking analysis was used to obtain the details of binding mode, essential amino acid residues of the binding site, and the intermolecular interactions of molecules (4 and 5) with the binding site of 3CL-pro protein. The molecules 4 and 5 show potent inhibition capacity against 3CLpro main protease through molecular docking analyses. In addition, MD simulation was performed to investigate the stability of binding interactions obtained through molecular docking analyses. The results obtained from this combined experimental and theoretical studies will provide key ideas to the future drug design approaches to develop potent molecules that inhibit the 3CL-pro site of SARS-CoV-2 domain. In a continuation of our previous work on the synthesis of new heterocyclic derivatives [23,24,25,26,27,28], we are interested in the synthesis of heterocyclic systems containing both quinoxaline and benzodiazepine moieties and the evaluation of their biological activities.
Materials and Methods
Synthesis
To a suspension of imine (4 mmol) in 40 mL of absolute ethanol, pyridinecarbaldehyde (6 mmol) and 3 drops of trifluoroacetic acid are added. The mixture is heated at reflux during 3 h. After cooling, the precipitate formed was filtered off under vacuum and the residue chromatographed on silica gel (hexane/ethyl acetate = 50:50 (v/v)) to afford two new compounds 4 and 5, respectively (Scheme 1).

Synthetic route to compounds 4 and 5 reported in this manuscript
3-[(2Z)-3-(2-pyridylmethyl)-1,2,3,4-tetrahydroquinoxalin-2-ylidene]-6-methyl-3,4-dihydro-2H-pyran-2,4-dione (4)
Yellow crystal, yield: 62%. 1H NMR (400 MHz, CDCl3) δ: 3.04 (dd, 1H, J = 14 Hz, J = 10.1 Hz), 3.31 (dd, 1H, J = 14 Hz, J = 2.3 Hz), 5.92 (dd, 1H, J = 10.1 Hz, J = 2.3 Hz), 5.15 (s, 1H, NH), 2.20 (s, 3H, CH3), 5.82(s, 1H, CH), 6.69–8.57 (m, 8Har), 15.70 (s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ: 19.94 (CH3), 37.49 (CH2), 50.90 (CH), 93.70 (Cq), 107.42 (CH), 115.70–118.87–119.37–121.72–128.09–124.16–136.69–149.14 (CHar), 123.36–136.21–158.26–163.65, 164.78 (Cq), 162.90–185.69(C = O).
6-Methyl-3-[(4-pyridin-2-yl)-2,3,4,5-tetrahydro-1H-1,5-benzodiazepin-2-yl]-3,4-dihydro-2H-pyran-2,4-dione (5)
Orange crystal, yield: 27%. 1H NMR (400 MHz, CDCl3) δ: 2.76 (dd, 1H, J = 12.7 Hz, J = 10.1 Hz), 4.85 (dd, 1H, J = 12.7 Hz, J = 2.5 Hz), 5.21 (dd, 1H, J = 10.1 Hz, J = 2.5 Hz), 5.79 (s, 1H, NH), 2.20 (s, 3H, CH3), 5.83(s, 1H, CH), 7.07–8.61 (m, 8Harom), 15.70 (s, 1H, NH). 13C NMR (100 MHz, CDCl3): 19.92 (CH3), 36.34 (CH2), 65.38 (CH), 96.10 (Cq), 107.10 (CH), 120.27–121.52–121.53–121.56–124.84–128.43–137.19–148.43 (CHar), 124.73–140.71–159.72–163.08, 164.10 (Cq), 172.26–184.75(C = O).
Single-Crystal X-ray Diffraction
Suitable crystals of 4 and 5 were used for the X-ray crystallographic analysis. The X-ray intensity data were measured on a Bruker Smart APEX CDD system equipped with a graphite monochromator and a Mo-Kα fine-focus sealed tube (λ = 0.71073 Å). Full spheres of data were collected under control of the APEX3 program [29] and reduced to F2 values with SAINT [29] which also performed least squares refinement of the unit cell parameters using ca. 9900 reflections drawn from the full dataset. An empirical absorption correction and merging of equivalent reflections was performed with SADABS [30], and the structures were solved by dual space methods [30]. The structure models were refined with full-matrix, least squares methods [31].
Hirshfeld Surface and ESP Analyses
The Hirshfeld surface and fingerprint plots of 4 and 5 were obtained using the Crystal Explorer 3.0 package [31].The dnorm plots were mapped with color scale range − 0.53 au (blue) and 1.25 au (red). The red spots on the Hirshfeld surface specify the interactions that involved hydrogen bonding. The 2D fingerprint plots were displayed using the expanded range 0.6–2.8 Å. The electrostatic potential surfaces (EPS) of 4 and 5 were determined using the DFT method at the B3LYP/6–31 + G(d,p) level of theory using the Gaussian 16 package [32]. In the EPS, the red region corresponds to hydrogen acceptors, while the blue one corresponds to hydrogen donors.
Theoretical Details
The geometry optimization of the ground states (GS) of 4 and 5 has been achieved with DFT method at the B3LYP/6–31 + G(d,p) level of theory as implemented in the Gaussian 16 package [32]. The X-ray crystallographic coordinates of 4 and 5 were used as starting input coordinates in the DFT calculations without any constraints. The GSs minima were confirmed by the frequency calculations at the same levels of theory (i.e., B3LYP/6–311 + + G(d,p)). Imaginary frequencies show positive values, which indicate that the optimized geometries are true minima. The solvent effects were considered using the polarizable continuum model (PCM) [33]. Natural bond orbital calculations were performed at the same level of theory from the optimized structures. The optimized structures, frontier molecular orbitals, and ESP were visualized using Gaussian View 6 [32].
Molecular Docking Study
In the present study, we have performed molecular docking studies to explore the interaction modes of ligands with the main protease of interest found in SARS CoV-2 3CLpro (3C-like protease). The 3D structure of the enzyme in complex with a covalent peptide N3 was retrieved from the Protein Data Bank (PDB ID: 6LU7) (https://www.rcsb.org/) [34]. The input files for molecular docking of the target enzyme and the studied inhibitors were carried out by using AutoDock tool 1.5.6 (http://autodock.scripps.edu/) according to the protocol as described by the Rizvi et al. [35]. In this case, the co-crystallized ligands and water molecules were removed from the selected enzyme and then protonated by adding polar hydrogen using the same program. Charges to the protein and compounds were assigned using the Kollman united atom and Gasteiger charge method, respectively. A grid box with a size of x = 26, y = 54, and z = 32 and center of x = − 9.768, y = 11.436, and z = 68.904 was defined to cover the N3 binding pocket (substrate binding site) in the 3CLpro enzyme [34]. AutoDock Vina software [36] was used to check the binding affinity (E in Kcal/mol) leading to the best poses of the studied ligands. The N3 peptide was used a control which was extracted from the crystal structure and redocked to the 3CLpro at the substrate binding region. The root mean square deviation (RMSD) obtained after super imposition of the redocked structure on the original co-crystalized structure was only 0.73 Å which is less than 2 Å. As a result, the validity of docking parameters are reasonable in order to predict the binding mode of compounds under study. The number of docked poses generated for each compound at the active pocket of 3CLpro is 100 and subsequently ranked according to binding energy. The lowest binding energy pose was considered the fittest and complexed with receptor for analysis. The molecular interactions and binding modes of the top poses were visually examined using the Discovery Studio software [37].
Molecular Dynamics Simulations
Molecular dynamics studies were conducted to determine dynamics of the complex and get affirmation on the drug binding mode stability in the specified simulation time. The drug parameters were defined by the general AMBER force field (GAFF) in the Antechamber module of AMBER20 [38]. The missing hydrogen atoms were added to the protein via TLeap. In case of protein, parameters were generated using the ff14SB force field [39]. Sodium ions were added to ensure neutrality of the system. The MD simulation was performed explicitly in TIP3P water box. Prior to running production phase, the system protein and compounds were energy minimized using 1000 steps of steepest descent and conjugate gradient. Gradual heating of the systems was then achieved up to 310 K in NVT ensemble [40]. The systems were equilibrated for 1 ns, and finally 100 ns of MD simulations was performed through NPT ensemble [41]. For long-range electrostatic interactions, the particle mesh Ewald technique was used [42]. The SHAKE algorithm was applied to consider constrain bonds involving hydrogen atoms [43]. CPPTRAJ module [44] of AMBER was used to calculate RMSD [45] and root mean square fluctuations (RMSF) [46].
MMPBSA Thermodynamic Estimation
Binding free energy of the compounds to the receptor macromolecule (3CLpro) was done to get an affirmation on the binding stability of the compounds. This was accomplished through molecular mechanics generalized Poisson Boltzmann surface area (MMPBSA) method [40]. This method is widely employed, robust, and reliable analytical tool. To carry out this, AmberTools18 MMPBSA.py script was used to analyze binding free energy of selected MD snapshots [40]. Briefly, binding free energy is calculated via the following equation:
Results and Discussion
Description of the Crystal Structure
Description of the Crystal Structure of 4
The crystal structure of 4 is depicted in Fig. 1.

Perspective view of 4 with labeling scheme and 50% probability ellipsoids. The intramolecular hydrogen bond is shown by a dashed line
The N1/C6/C1/N2/C8/C7 ring is not planar, and a puckering analysis yielded the parameters Q = 0.358(3) Å, θ = 60.8(5)°, and φ = 325.3(6)°. The dihydropyrandione and the 2-pyridyl rings are inclined to the C1···C6 ring by 20.20(8) and 24.04(9)°, respectively. One notable feature is the long C8—C15 bond distance (1.421(4) Å) and the short C8—N2 bond distance (1.326(4) Å). Also, the sums of the interbond angles about N2, C8, and C15 are all 360° within experimental error suggesting that all three atoms are predominantly sp2 hybridized. Similar shortening of the corresponding C—N bond and lengthening of the C—C bond has been found in a number of related molecules although not to the extent see here [47,48,49,50,51,52]. In the crystal, \(\begin{array}{cc}\mathrm{N}1-\mathrm{H}1& \mathrm{O}3\end{array}\) hydrogen bonds (Table 1) form chains of molecules extending along the b-axis direction. These are connected in pairs by \(\begin{array}{cc}\mathrm{C}13-\mathrm{H}13& \mathrm{O}1\end{array}\)· and \(\begin{array}{cc}\mathrm{C}20-\mathrm{H}20\mathrm{B}& \mathrm{O}1\end{array}\)· hydrogen bonds (Table 1) with the resulting ribbons associated through slipped π-stacking interactions (centroid···centroid = 3.6349(18) Å, dihedral angle = 3.80(15)°) between the dihydropyrandione and 2-pyridyl rings (Figs. 2 and 3).

Packing of 4 viewed along the a-axis direction with N—H···O and C—H···O hydrogen bonds shown, respectively, by blue and black dashed lines. The π-stacking interactions are shown by orange dashed lines

Packing of 4 viewed along the b-axis direction with C—H···O hydrogen bonds shown by black dashed lines. The π-stacking interactions are shown by orange dashed lines
Description of the Crystal Structure of 5
The crystal structure of 5 is depicted in Fig. 4.

Perspective view of 5 with labeling scheme and 50% probability ellipsoids. Only the major orientation of the disordered dihydropyran ring is shown
In 5, the dihedral angle between the C1···C6 and C10···C14/N3 rings is 43.36(6)°. The pendant dihydropyran ring shows a 10% disorder with the minor component rotated by 174.6(4)° from the orientation of the major component. The orientation of both components is largely determined by intramolecular \(\begin{array}{cc}\mathrm{N}2-\mathrm{H}2\mathrm{A}& \mathrm{O}2\end{array}\)· or \(\begin{array}{cc}\mathrm{N}2-\mathrm{H}2\mathrm{A}& \mathrm{O}3\mathrm{A}\end{array}\)· hydrogen bonds (Table 2 and Fig. 4). A puckering analysis [38] of the major orientation of the dihydropyran ring gave the parameters Q = 0.127(2) Å, θ = 108.0(8)°, and φ = 79.6(8)°, while for the 7-membered ring, the parameters are Q(2) = 0.8888(13) Å, Q(3) = 0.2070(13) Å, φ(2) = 201.03(8)°, and φ(3) = 293.9(4)°. In the crystal, inversion-related molecules form dimers through \(\begin{array}{cc}\mathrm{N}1-\mathrm{H}1& \mathrm{O}2\end{array}\)· hydrogen bonds (Table 2 and Fig. 5) which are associated along the b-axis direction by slipped π-stacking interactions between the pendant pyridyl and dihydropyran rings (centroid···centroid = 3.833(3) Å, dihedral angle = 14.51(2)°).

Packing in 5 viewed along the a-axis direction with N—H···O hydrogen bonds and slipped π-stacking interactions shown, respectively, by blue and green dashed lines
Hirshfeld Surface Studies
The Hirshfeld surfaces of 4 and 5 display the regions of intermolecular interactions (intercontacts) that may be established in the crystalline environment (Fig. 6). The red spots over the Hirshfeld surface indicate the interactions that involve hydrogen bonding. For both compounds, the hydrogen bonds are formed between donor and acceptor atoms. Figure 6 shows a strong hydrogen bond between the NH group of piperazine and the 1,4-diazepine of 4 and 5 to form one side of the basic unit and the lone pair of the oxygen atom of the pyran-2,4-dione of the closest unit from other side at distances of 1.92 and 2.48 Å, respectively (Fig. 6). In the EPS of 4 and 5, the negative electrostatic potential appears in the red region over the oxygen atom of the oxo group confirming its role as a hydrogen bond acceptor (Fig. 7).

dnorm of 4 and 5 mapped on the Hirshfeld surface for visualizing the intermolecular contacts

EPS of 4 and 5 obtained at the B3LYP/6–31 + G(d,p) level of theory
The intercontacts over the Hirshfeld surfaces of 4 and 5 are quantified using 2D fingerprint plots (Fig. 8). The major contributions are found between H···H at 49.1 and 46.2% for 4 and 5, respectively. Other interesting intercontacts with contribution ranges 15–20% are found between \(\begin{array}{cc}\mathrm{C}& \mathrm{H}/\mathrm{H}\end{array}\)· and \(\begin{array}{cc}\mathrm{O}& \begin{array}{cc}\mathrm{H}/\mathrm{H}& \mathrm{O}\end{array}\end{array}\).

Fingerprint plots of all (right) and highest interatomic interactions of 4 and 5
DFT Results
The optimized geometries of 4 and 5 were obtained at the B3LYP/6–31 + G(d,p) level of theory in a polarizable continuum solvent model. Some of the optimized and X-ray bond lengths, bond angles, and torsion angles are shown in Table 3. The superposition of the optimized and X-ray geometries shows an excellent agreement between the experimental and calculated Z-matrix coordinates (Fig. 9). This agreement is confirmed by the relatively good correlation between the calculated and X-ray geometrical parameters. The correlation coefficients between bond length, bond angles, and torsion angles of the optimized and X-ray structures are in the 91–99.50% range.

Superposition between X-ray and optimized geometries of 4 and 5
The intramolecular and delocalization of electron density in 4 and 5 are investigated by analyzing the natural bond orbital (NBO) outputs. The larger is the value of the mean energy of hyper conjugative interaction E(2), the more is the interaction between electron donors and acceptors. In other word, the more donation tendency from electron donor atom to electron acceptor atom, the greater is the extent of conjugation over the entire system. The filled bonding or lone pair orbitals may act as an electron donor, while the empty anti-bonding or non-bonding orbital may act as an electron acceptor. These interactions may affect the strength of bonds involving the atoms in question. The NBO calculations have been carried to investigate all possible interactions between “filled” (donor) Lewis-type NBOs and “empty” (acceptor) non-Lewis NBOs.
For 4, the strongest interaction with a stabilization energy of 284.06 kcal mol−1 is established between the lone pair LN (1) Lewis NBO of C18 and non-Lewis anti-bond NBO π*(N6-C9). The lone pair LP(1) NBO of N4 conjugated with the LN(1) NBO of C18 leads to a strong stabilization of energy of 64.92 kcal/mol. For 5, a strong interaction with stabilization energy of 304.93 kcal mol−1 was found due to electron density transfer from the lone pair LN(1) NBO of C15 to the conjugated π*(N3-C6) NBO anti-bond. The lone pair LP(1) of N1 conjugated with the LN(1) of C15 leads to a strong stabilization of energy of 83.13 kcal/mol. These strong stabilization energies denote the larger delocalization of 4 and 5.
The chemical reactivity of 4 and 5 is emphasized by the analysis of the frontier molecular orbitals HOMO and LUMO. The HOMO, LUMO, and gap energies are shown in Fig. 10. The FMOs indicate the intramolecular charge transfer (ICT) from the decahydro-1H-benzo[b][1,4]diazepine and decahydroquinoxaline moieties in 4 and 5 (HOMO) to dihydro-2H-pyran-2,4(3H)-dione (LUMO), respectively. The band gap energy values of 4 and 5 are 3.56 and 3.48 eV, respectively. The lower gap energy of 5 compared with 4 may indicate a higher activity of the former compared with the latter (Fig. 10).

Energetic profile of FMOs of 4 (left) and 5 (right) obtained at the B3LYP/6–31 + G(d,p) level of theory
Molecular Docking Analysis
Molecular docking simulations were carried out for 4 and 5 with the SARS CoV-2 main protease 3CLpro enzyme. Table 4 summarizes the docking results with their corresponding binding affinity (E in Kcal/mol) and types of bond interactions. Based on the energy calculations, 4 and 5 exhibited strong interactions with the binding site of 3CLpro enzyme.
Compound 4 was docked into the active pocket of the 3CLpro with a binding energy of − 8.6 kcal/mol. Two conventional hydrogen bonds are formed, one by the carbonyl group (C = O) with the negatively charged glutamic amino acid (GLU166) and the other by the positively charged histidine amino acid (HIS44) with the pyridine group of 4 (Fig. 11). In addition, histidine (HIS44) forms one π-cation bond with the benzyl group, while two π-alkyl bonds from the hydrophobic methionine (MET49) interact with the benzyl ring, and cysteine (CYS145) interacts with the pyridine ring. Furthermore, one π-sulfur bond was observed between the sulfur atom in methionine (MET165) and the benzyl group of 4 (see Fig. 11).

3D and 2D representations of compound 4 in the binding pocket of main protease enzyme. In case of 3D, hydrogen bonding in terms of acceptor and donor regions is shown along with coloring on hydrophobicity basis. In the 2D structure, compound 4 is shown via gray stick, while residues are presented by different color disc each presenting specific interaction
Similarly, 5 has a good binding affinity (E = − 8 kcal/mol) towards the main protease 3CLpro receptor with the formation of one hydrogen bond to cysteine (CYS145) with the carbonyl group (C = O) of 5. Moreover, the pyridine ring of 5 forms two π-alkyl bonds to the hydrophobic methionines (MET49 and MET165) (Fig. 12). Two other π-alkyl bonds were observed between the positively charged histidines (HIS172 and HIS163) and the methyl group 5. One π-cation bond to the positively charged histidine amino acid (HIS41) from the pyridine ring of 5 (see Fig. 12).

3D and 2D representations of 5 in the binding site of the main protease 3CLpro enzyme through molecular docking analysis. In case of 3D, hydrogen bonding in terms of acceptor and donor regions is shown along with coloring on hydrophobicity basis. In the 2D structure, compound 5 is shown via gray stick, while residues are presented by different color disc each presenting specific interaction
MD Analysis
MD simulations were performed for 4 and 5 with the SARS CoV-2 3CLpro enzyme to investigate their stability and dynamic properties at the atomic level. The best poses of 4 and 5 obtained from the molecular docking studies were simulated at 100 ns. To assess the conformational stability of the 3CLpro enzyme during the MD simulation, the RMSD was calculated as a function of time for 3CLpro-4 (or 5) complexes (Fig. 13A). The RMSD for both 4 and 5 were well equilibrated at around 2–2.5 Å and remained constant during the 100 ns simulation with no significant differences observed. From Fig. 13A, it can be seen that RMSD value of 3CLpro-5 behaves with a bit higher RMSD (though consistent) showing higher structural flexibility of the system as compared to that of the 3CLpro-4 complex. Those results imply that the protein with 4 shows relatively greater stability than the 3CLpro-5 complex. Interestingly, the RMSD values of both systems reach equilibrium under the RMSD after initial 10 ns, which indicate that the complexes are stable and the difference between them is not significant. In addition, residue level fluctuations of the systems were measured using the RMSF, which maintained a stable RMSF value over time (Fig. 13B). The RMSD values are within highly acceptable range of 2 2–2.5 Å. It also indicated the stability of the residues of the 3CLpro enzyme.

Plots of RMSD and RMSF of complexes over 100 ns of MD simulation: A RMSD vs time and B RMSF vs time
The MD simulation analysis of the docked 4 and 5 validated the stability of 3CLpro-4 (or 5) complexes. This stability was confirmed by the previous studies, which reported that the docked molecule interaction with the binding site in the main protease 3CLpro enzyme occurs around the active sites containing HIS41, CYS145, MET49, MET165, HIS172, and GLU166 amino acids [53, 54].
Binding Free Energy Calculation
MMPBSA binding free energy is an inexpressive, dynamic, and a useful tool in estimation of atomic level interactions between therapeutic compounds to its respective biological macromolecule. It can be utilized as post docking validation assay and reconfirm ligand binding and stability at the docked position of the biological target and is also helpful in predicting key contributing residue in ligand recognition. The entropy effect is computationally expensive therefore not considered during binding energy estimation. Both the compounds presented highly favorable binding free energies. The different binding energy components of both systems are provided in Table 5. The net binding energy of both complexes is an indication of high intermolecular affinity and stability.
Conclusions
The present study attempts to explore two new molecules as SARS CoV-2 3CLpro complexes for treatment of COVID-19. Herein we highlight the significant interaction of these compounds with 3CLpro enzyme that can be rationalized to activate the receptor and hence exert an antiviral effect. The refined structures (4 and 5) were investigated through X-ray crystallography, whereas the closest intercontact interactions between 4 and 5 units were studied identified by the analysis of the Hirschfeld surface and the electrostatic potential surfaces (EPS). Furthermore, the binding affinity and stability of these compounds to the binding site of SARS-CoV-2 3CLpro receptor were investigated by molecular docking and MD simulations. For both studied molecules, significant binding affinity was demonstrated through molecular docking studies with various types of interactions. Following the computational procedures, the substrates were exposed to MD simulations to determine the rigidity of the protein structure, fluctuations created during interactions, and structural stability collectively by calculating RMSD and Rg parameters. Notably, 4 exerted minimal conformational changes and negligible fluctuations and was structurally stabilized with 3CLpro enzyme of COVID-19. This was further validated by estimation of MMPBSA binding free energy which confer strong evidence in support of high affinity of both designed compounds to the 3CLpro target. In accordance with this, the derivative implied a strong interaction with the receptor which apt the possibility of serving an effective antiviral compound.
References
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., & Pei, Y.-Y. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579, 265–269.
Wu, J. T., Leung, K., & Leung, G. M. (2020). Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. The Lancet, 395, 689–697.
Hui, D. S., Azhar, E. I., Madani, T. A., Ntoumi, F., Kock, R., Dar, O., Ippolito, G., Mchugh, T. D., Memish, Z. A., & Drosten, C. (2020). The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. International Journal of Infectious Diseases, 91, 264–266.
Chan, J.F.-W., Yuan, S., Kok, K.-H., To, K.K.-W., Chu, H., Yang, J., Xing, F., Liu, J., Yip, C.C.-Y., & Poon, R.W.-S. (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. The Lancet, 395, 514–523.
Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., & Huang, C.-L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579, 270–273.
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., & Gu, X. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395, 497–506.
Cui, J., Li, F., & Shi, Z.-L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17, 181–192.
Van Boheemen, S., de Graaf, M., Lauber, C., Bestebroer, T. M., Raj, V. S., Zaki, A. M., Osterhaus, A. D., Haagmans, B. L., Gorbalenya, A. E. & Snijder, E. J. (2012). Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio, 3, e00473–12.
Wu, X. D., Shang, B., Yang, R. F., Hao, Y., Hai, Z., Xu, S., Ji, Y. Y., Ying, L., Di, Wu., & Y. & Lin, G. M. . (2004). The spike protein of severe acute respiratory syndrome (SARS) is cleaved in virus infected Vero-E6 cells. Cell research, 14, 400–406.
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C.-L., Abiona, O., Graham, B. S., & McLellan, J. S. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367, 1260–1263.
Skariyachan, S., Challapilli, S. B., Packirisamy, S., Kumargowda, S. T., & Sridhar, V. S. (2019). Recent aspects on the pathogenesis mechanism, animal models and novel therapeutic interventions for Middle East respiratory syndrome coronavirus infections. Frontiers in microbiology, 10, 569.
Mustafa, S., Balkhy, H., & Gabere, M. N. (2018). Current treatment options and the role of peptides as potential therapeutic components for Middle East Respiratory Syndrome (MERS): A review. Journal of infection and public health, 11, 9–17.
Hui, D. S., Azhar, E. I., Kim, Y.-J., Memish, Z. A., Oh, M.-D., & Zumla, A. (2018). Middle East respiratory syndrome coronavirus: Risk factors and determinants of primary, household, and nosocomial transmission. The Lancet Infectious Diseases, 18, e217–e227.
Ziebuhr, J. (2004). Molecular biology of severe acute respiratory syndrome coronavirus. Current opinion in microbiology, 7, 412–419.
Rota, P. A., Oberste, M. S., Monroe, S. S., Nix, W. A., Campagnoli, R., Icenogle, J. P., Penaranda, S., Bankamp, B., Maher, K., & Chen, M.-H. (2003). Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science, 300, 1394–1399.
Drosten, C., Günther, S., Preiser, W., Van Der Werf, S., Brodt, H.-R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L., & Fouchier, R. A. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England journal of medicine, 348, 1967–1976.
Marra, M. A., Jones, S. J., Astell, C. R., Holt, R. A., Brooks-Wilson, A., Butterfield, Y. S., Khattra, J., Asano, J. K., Barber, S. A., & Chan, S. Y. (2003). The genome sequence of the SARS-associated coronavirus. Science, 300, 1399–1404.
Ruan, Y., Wei, C. L., Ling, A. E., Vega, V. B., Thoreau, H., Thoe, S. Y. S., Chia, J.-M., Ng, P., Chiu, K. P., & Lim, L. (2003). Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. The Lancet, 361, 1779–1785.
Skariyachan, S., Gopal, D., Chakrabarti, S., Kempanna, P., Uttarkar, A., Muddebihalkar, A. G., & Niranjan, V. (2020). Structural and molecular basis of the interaction mechanism of selected drugs towards multiple targets of SARS-CoV-2 by molecular docking and dynamic simulation studies-deciphering the scope of repurposed drugs. Computers in biology and medicine, 126, 104054.
Teli, D. M., Shah, M. B., & Chhabria, M. T. (2021). In silico screening of natural compounds as potential inhibitors of SARS-CoV-2 main protease and spike RBD: Targets for COVID-19. Frontiers in molecular biosciences, 7, 429.
Cubuk, H., & Özbİl, M. (2021). Comparison of clinically approved molecules on SARS-CoV-2 drug target proteins: A molecular docking study. Turkish journal of chemistry, 45, 35–41.
Fakhar, Z., Khan, S., AlOmar, S. Y., Alkhuriji, A. & Ahmad. (2021). A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Scientific reports, 11, 1–15.
Sebhaoui, J., El Bakri, Y., Lai, C.-H., Karthikeyan, S., Anouar, E. H., Mague, J. T., & Essassi, E. M. (2020). Unexpected synthesis of novel 2-pyrone derivatives: Crystal structures, Hirshfeld surface analysis and computational studies. Journal of Biomolecular Structure and Dynamics, 38, 1–19.
El Bakri, Y., Karthikeyan, S., Anouar, E. H., Sebhaoui, J., Ben Ali, A., El Ghayati, L., Essassi, E. M., & Mague, J. T. (2020). A newly synthesized 6-methyl-7 H, 8 H, 9 H-[1, 2, 4] triazolo [4, 3-b][1, 2, 4] triazepin-8-one for potential inhibitor of adenosine A1 receptor: A combined experimental and computational studies. Journal of Biomolecular Structure and Dynamics, 38, 3578–3586.
El Bakri, Y., Subramani, K., Ben-Yahya, A., & Essassi, E. M. (2020). Synthesis, spectroscopic characterizations, DFT, molecular docking and molecular dynamics simulations of a novel 2-methyl-3H-benzimidazolo [1, 2-b][1, 2, 4] triazepin-4 (5H)-one. Journal of Molecular Structure, 1202, 127317.
Abad, N., El Bakri, Y., Lai, C.-H., Karthikeyan, S., Ramli, Y., Ferfra, S., Mague, J. T. & Essassi, E. M. (2020). Insights into the crystal structure of two newly synthesized quinoxalines derivatives as potent inhibitor for c-Jun N-terminal kinases. Journal of Biomolecular Structure and Dynamics, 1–18.
El Bakri, Y., Marmouzi, I., El Jemli, M., Karthikeyan, S., Harmaoui, A., Faouzi, M. E. A., Mague, J. T., & Essassi, E. M. (2019). Synthesis, biological activity and molecular modeling of a new series of condensed 1, 2, 4-triazoles. Bioorganic chemistry, 92, 103193.
El Bakri, Y., Marmouzi, I., Sayah, K., Ramli, Y., Faouzi, M. E. A., Essassi, E. M., & Mague, J. T. (2018). Potential antidiabetic activity and molecular docking studies of novel synthesized 3.6-dimethyl-5-oxo-pyrido [3, 4-f][1, 2, 4] triazepino [2, 3-a] benzimidazole and 10-amino-2-methyl-4-oxo pyrimido [1, 2-a] benzimidazole derivatives. Journal of molecular modeling, 24, 179.
Bruker, A. & Saint, A. (2008). Inc., Madison, WI, 2004 Search PubMed;(b) GM Sheldrick. Acta Crystallogr., Sect. A: Fundam. Crystallogr, 64 112
Sheldrick, G. M. (2015). Crystal structure refinement with SHELXL. Acta Crystallographica Section C: Structural Chemistry, 71, 3–8.
Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). CrystalExplorer17. University of Western Australia.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, J. A., Peralta, Jr., J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin,K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B. & Fox, D. J. (2016). Gaussian 16, Revision A.03. Gaussian, Inc., Wallingford CT.
Tomasi, J., Mennucci, B., & Cammi, R. (2005). Quantum mechanical continuum solvation models. Chemical Reviews, 105, 2999–3094.
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L. & Peng, C. (2020). Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature, 1–5.
Rizvi, S. M. D., Shakil, S., & Haneef, M. (2013). A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. Excli Journal, 12, 831.
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31, 455–461.
BIOVIA, D. S. & Dsme, R. (2015). San Diego: Dassault systèmes. Release.
Case, D.A., Aktulga, H.M., Belfon, K., Ben-Shalom, I.Y., Brozell, S.R., Cerutti, D.S., Cheatham, T.E., Cruzeiro, III, V.W.D., Darden, T.A., Duke, R.E., Giambasu, G., Gilson, M.K., Gohlke, H., Goetz, A.W., Harris, R., Izadi, S., Izmailov, S.A., Jin, C., Kasavajhala, K., Kaymak, M.C., King, E., Kovalenko, A., Kurtzman, T., Lee, T.S., LeGrand, S., Li, P., Lin, C., Liu, J., Luchko, T., Luo, R., Machado, M., Man, V., Manathunga, M., Merz, K.M., Miao, Y., Mikhailovskii, O., Monard, G., Nguyen, H., O’Hearn, K.A., Onufriev, A., Pan, F., Pantano, S., Qi, R., Rahnamoun, A., Roe, D.R., Roitberg, A., Sagui, C., Schott-Verdugo, S., Shen, J., Simmerling, C.L., Skrynnikov, N.R., Smith, J., Swails, J., Walker, R.C., Wang, J., Wei, H., Wolf, R.M., Wu, X., Xue, Y., York, D.M., Zhao, S., & Kollman P.A. (2021). Amber 2021. University of California, San Francisco.
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11, 3696–3713.
Abro, A., & Azam, S. S. (2016). Binding free energy based analysis of arsenic (+ 3 oxidation state) methyltransferase with S-adenosylmethionine. Journal of Molecular Liquids, 220, 375–382.
Ahmad, S., Raza, S., Uddin, R., & Azam, S. S. (2017). Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. Journal of Molecular Graphics and Modelling, 77, 72–85.
Plimpton, S., Pollock, R. & Stevens, M. (1997). Particle-mesh Ewald and rRESPA for parallel molecular dynamics simulations. PPSC.
Kräutler, V., Van Gunsteren, W. F., & Hünenberger, P. H. (2001). A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry, 22, 501–508.
Roe, D. R., & Cheatham, T. E., III. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9, 3084–3095.
Maiorov, V. N., & Crippen, G. M. (1994). Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. Journal of molecular biology, 235, 625–634.
Elfiky, A. A. (2021). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 39, 3194–3203.
Touzani, R., Ben-Hadda, T., Elkadiri, S., Ramdani, A., Maury, O., Le Bozec, H., Toupet, L., & Dixneuf, P. H. (2001). Solution, solid state structure and fluorescence studies of 2, 3-functionalized quinoxalines: Evidence for a π-delocalized keto-enamine form with N-H··· O intramolecular hydrogen bonds. New journal of chemistry, 25, 391–395.
Kaitner, B., Jovanovski, G., & Janev, I. (1992). Structure of 2,3-diphenacylquinoxaline. Acta Crystallographica Section C, 48, 129–131.
Seki, T., Iwanami, Y., Kuwatani, Y., & Iyoda, M. (1997). Heterocycles structurally influenced by a side chain. X. Effect of temperature and side chain on the imine-enamine tautomerism in the quinoxalinone and pyridopyrazinone systems. Journal of Heterocyclic Chemistry, 34, 773–780.
Prokop, P., Richter, R., & Beyer, L. (1999). Zur Reaktion von 2,4-Dioxo-4-ferrocenyl-butansäureethylester mit primären aromatischen Aminen / On the reaction of ethyl 2,4-dioxo-4-ferrocenyl-butanoate with primary aromatic amines. Zeitschrift für Naturforschung B, 54, 849–857.
Liu, Y., Lu, Z.-F., Shen, Y.-M., & Xu, J.-H. (2005). 3-Benzoylmethylene-1-methyl-3,4-dihydroquinoxalin-2(1H)-one. Acta Crystallographica Section E, 61, o2753–o2754.
Luo, Y., Tao, F., Liu, Y., Li, B., & Zhang, G. (2006). Intramolecular amidation — An efficient synthesis of 3-aryl-2-quinolinones. Canadian Journal of Chemistry, 84, 1620–1625.
Macchiagodena, M., Pagliai, M., & Procacci, P. (2020). Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chemical Physics Letters, 750, 137489.
Nukoolkarn, V., Lee, V. S., Malaisree, M., Aruksakulwong, O., & Hannongbua, S. (2008). Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. Journal of theoretical biology, 254, 861–867.
Funding
The authors gratefully acknowledge Mohammed V University (Morocco) for the financial assistance and facilitation to facilitate this study. The support of Tulane University for the Tulane Crystallography Laboratory is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
CCDC 2056720, and 1884597 contains the supplementary crystallographic data for 4 and 5, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Rights and permissions
About this article

Cite this article
El Bakri, Y., Anouar, E.H., Ahmad, S. et al. Synthesis and Identification of Novel Potential Molecules Against COVID-19 Main Protease Through Structure-Guided Virtual Screening Approach. Appl Biochem Biotechnol 193, 3602–3623 (2021). https://doi.org/10.1007/s12010-021-03615-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-021-03615-8