
Abstract
Cryopreservation abilities of dental tissue-derived mesenchymal stromal cells (DMSCs) including dental pulp stem cells (DPSCs) and dental follicle stem cells (DFSC) play an important role in the applications of these cells in clinical settings. In this context, we checked whether storage at − 80 °C in 10% DMSO for a longer period has any adverse effect on the functionality and genetic stability. We carried our studies on DPSC and DFSC samples that were revived after a maximum of 5 years of cryopreservation. We observed that even after long-term uncontrolled freezing at − 80 °C, these cells survived and proliferated efficiently. The assessment was made based on their post-thaw morphology, immunophenotypes, differentiation potential, growth kinetics, and genetic features. These cells retained the expression of stemness markers, differentiation ability and maintained their normal karyotype. Our results indicated no significant morphological or immunophenotypic differences between the cryopreserved DMSCs and the fresh DMSCs. Our study implies that mesenchymal stromal cells derived from the dental tissue origin are very robust and do not require any sophisticated preservation protocols. Thus, these can be an ideal source for research, stem cell banking, as well as successful clinical applications in tissue engineering and cell-based therapeutics.

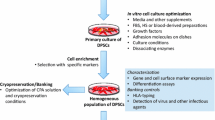
Schematic diagram showing the cryopreservation of DMSCs by uncontrolled freezing at -80 c has no adverse effects on their functionality and genetic stability
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the modern era of regenerative medicine, stem cell–based therapy has revolutionized conventional therapeutic setups. Various in vitro up-scaling strategies [1,2,3,4] have been developed to achieve a substantial number of cells required for any cell-based therapy. However, prolonged in vitro culturing of mesenchymal stromal cells leads to the introduction of genetic/epigenetic instability which can alter their characteristics and functional properties resulting in compromised clinical benefits [5, 6]. Short-term preservation of stem cells at 4 °C may be promising during clinical trials, but maintaining cell viability at this temperature is time-bound, that ranges from 2 to 4 h, depending upon the different storage conditions and longer duration significantly hampers cells viability and is not useful [7, 8]. Therefore, for quantitative and qualitative stem cell-based therapies, long-term storage of the cells at early passages can be a beneficial factor.
Current techniques employed for the storage of cells include conventional cryopreservation methods that employ the addition of cryoprotectant, programmed slow freezing, and rapid freezing (vitrification) [9,10,11]. The conventional method utilizing cryoprotectants lowers the freezing point of the medium containing cells, thus allowing the intracellular fluid to move out of the cells at the rate that is neither too slow to dehydrate cells and disrupt the integrity of the cell membrane nor too fast to form ice-crystal inside the cells [12]. Cryopreservation enables long-term as well as on-site storage of stem cells, facilitating their direct administration at the time of need [13]. Different responses to cryopreservation have been observed for various types of stem cell, and these variabilities are attributed to differences in the cellular properties like cell size, cell membrane permeability, Arrhenius activation energy (that influence the temperature dependence of cell membrane permeability to water and different cryoprotectant agents), and osmotic tolerance of cells [14,15,16]. Since the varied cryobiological responses make it difficult to adopt a single cryopreservation method universally, thus, it is essential to strengthen the need for practicing a wider range of cryopreservation techniques designed for specific cell types. For instance, a study demonstrated that the vitrification method was more suitable for cryopreservation of human embryonic stem cells (hESCs) in comparison to conventional cryopreservation in terms of post-thawed attachment rate and recovery of hESC [11]. Similarly, dimethyl sulfoxide (DMSO) along with the fetal bovine serum is widely used to protect the cells from rapid freezing and thawing cycles [12, 17]. A recent study on the impact of varying rates of cooling and thawing on the quality of cryopreserved cells revealed that when T cells are frozen in DMSO at slow cooling, subsequent warming rate does not affect post-thaw cell viability [18]. However, DMSO can have adverse effects on cell viability and functionality since it is cell membrane permeable. The cryopreservation protocols should be able to overcome the post-thaw issues like poor cell recovery, reduced cell viability, and the hampered proliferation rate of recovered cells [4]. A growing number of evidence support the use of 10% DMSO during cryopreservation for successful post-thaw cell viability and differentiation of MSCs from adipose and dental pulp tissues [19]. The current cell-based therapeutic strategies that demand cryopreservation of progenitor cells for all autologous and many allogeneic transplantations can be fulfilled by biobanking [20]. The banking of the mesenchymal stromal cells utilizing cryopreservation technologies enables a constant supply of allogenic cells as “off the shelf’ therapeutic product to be used in patients immediately at the time of need [21, 22]. Successful cryopreservation of stem cells is a key factor that enables storage, the safe and convenient shipment of stocks of quality-controlled lines of stem cells between different geographic locations at a reasonable cost [23]. Other benefits of cryopreservation include the constant availability of adequate cells for a proper therapeutic dose, extensive quality testing of the cells to ensure the safety and suitability for transplantation by stem cell biobanking. In this regard, we have previously reported various DMSO-based cryopreservation protocols for the storage of dental pulp stem cells. We observed post-thaw viable stem cells with unaltered plating and differentiation efficiency after 1 year of storage at − 80 °C [24]. Based on the previous findings, the present study evaluates the effect of uncontrolled freezing at − 80 °C on post-thaw viability and multipotency of dental mesenchymal stromal cells after 5 years of cryopreservation. Our data may illustrate a promising long-term cryopreservation method for DMSCs that does not require the need for liquid nitrogen storage.
Methods
DMSC Cell Culture and Cryopreservation
Dental mesenchymal stromal cells used for the present study were the samples stored at − 80 °C, collected from previously recruited patients who visited oral health science center, PGIMER, OPD for tooth extraction under the advice of the consulting surgeon. Written consent was signed by the patient, and all the protocols were approved by the Institute Ethics Committee. Tooth extraction was carried out under local anesthesia, and proper sterile conditions were maintained during the procedure. Teeth were collected from patients and transported to tissue culture laboratory for stem cell isolation. The culture of DMSCs was obtained by explant culture method as reported before [24]. DMSCs were cultured in αMEM (Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (FBS) (HiMedia, India) and antibiotic cocktail (penicillin, streptomycin, and amphotericin B) (HiMedia, India). Cells were maintained in a humidified incubator at 37 °C with 5% CO2. The culture medium was replaced with fresh media every third day. For cryopreservation, the previously described protocol was followed [24]. Briefly, the DMSCs (1 × 106) were trypsinized and centrifuged at 2000 rpm for 2 min with FBS. The cell pellet was then washed with PBS followed by centrifugation. Thereafter, the cell pellet was resuspended in cryomedia comprising 10% DMSO (Sigma-Aldrich, USA) and 90% FBS and the cell suspension was quickly transferred into cryovial and directly stored at − 80 °C freezers.
Post-thaw Revival of DMSCs
DMSC stocks (Table 1) stored at − 80 °C were thawed by immediately transferring the vials to 37 °C for 2–3 min. Afterward, cells were quickly transferred to 5 ml α-MEM media in a 15-ml centrifuge tubes followed by 2 min centrifugation at 2000 rpm. After discarding the supernatant, complete α-MEM media with 10% FBS was added to resuspend the pellet. Cells were seeded into T-25 flasks and kept at 37 °C incubator with 5% CO2. Cells were regularly monitored under brightfield inverted microscope for their morphology, and media was replaced after every third day. Confluent cultures of these cryopreserved DMSCs were trypsinized and sub-cultured before using them in further experiments.
Phenotypic Characterization
The DMSCs after achieving 70 to 80% confluency were subjected to stemness phenotypic characterization. Briefly, cells were trypsinized using trypsin EDTA 0.25% (Sigma-Aldrich, USA). After centrifugation, the cell pellet was washed twice with phosphate-buffered saline solution (PBS) (HiMedia, India) and incubated with stem cell-specific antibodies viz. CD90, CD73, CD105, and negative antibodies including endothelial marker CD34 and hematopoietic marker CD45 (BioLegend, USA), directly conjugated to Fluorescein isothiocyanate (FITC) in dark for 30 min. Cells were then washed again with PBS and acquired in flow cytometry (Becton–Dickinson, FACS Calibur) and analyzed using FloJo.
Post-Thaw Cells Viability and Proliferation
MTT assay was performed for cell viability. Briefly, 5 × 103cells per well were plated in 96-well plates containing complete α-MEM media with 10% FBS for 48 h. Cultured media replaced with fresh media and MTT reagent (Sigma-Aldrich, USA) was added. Cells were incubated for 4 h followed by DMSO mediated cell lysis. Wells were read at 565 nm wavelength (Tecan, M7500 pro, Switzerland). Similarly, for cell proliferation, 5 × 103 cells were seeded in 96 wells in the presence of complete α-MEM medium and cultured for 48 h. This was followed by the addition of EZBlue dye (HiMedia, India) as per the manufacturer’s instructions. Reading was taken at a wavelength of 570 nm (Tecan ELISA reader cum spectrophotometer, Switzerland).
Cells Apoptosis Detection
Cryopreserved DMSCs were analyzed for Apoptosis and necrosis by using Annexin V apoptosis detection kit (BioLegend, USA). Briefly, cell culture flasks were seeded with an equal number of cells and, at 70% confluency, cells were trypsinized and processed further as per the manufacturers’ instructions. In the end, DMSCs were acquired in a flow cytometer (Becton–Dickinson, FACSCalibur, USA).
Cell Cycle Analysis
A total of 0.5 × 106 of cells were seeded in a T-25 culture flask. Cells were cultured for 48 h, trypsinized, washed with PBS, and fixed in 90% ethanol overnight at − 20 °C. The next day, cells were centrifuged at 3000 rpm for 5 min and washed with PBS. After washing, cells were incubated with RNAse/PI (molecular probes/Invitrogen, USA) solution for 10 min and then acquired in the flow cytometer (Becton– Dickinson, FACS Calibur). Distribution of cells in Go/G1, S, and G2/M phase was observed to correlate it with the growth status of the cells.
Colony Formation Assay
Single-cell suspension after trypsinization of each cell type was plated in six-well plates at a no. of 1 × 103 cells per well [25]. These cells were allowed to grow for 7 days under defined culture conditions with the change of culture media as per requirement. After 7 days, PBS washing was given to the cells before fixing them in 10% neutral buffered formalin. Cells were stained with crystal violet (0.1%), and colonies were observed under the microscope (Nikon Eclipse TS 100, USA). The number of cells greater than 50 was counted as a single colony.
Sphere Forming Assay
For sphere formation, 1000 cells were plated per well in ultra-low attachment 6-well plates (Corning, USA). Cells were observed regularly for sphere growth up to day 7. Images were captured with the inverted microscope (Nikon Eclipse TS100) on day 0, 3, 5, and 7. The average diameter was calculated by using a software named NIS-Elements D4.13.00. Five images per sample were used for calculating the diameter. The viability of spheres at day 7 was assessed by adding 2 μl of 1 mg/ml acridine orange (AO) and ethidium bromide (EtBr) (HiMedia, India) mixture prepared in PBS directly to the wells followed by 10–15 min incubation at 37 °C incubator with 5% CO2. Spheres were viewed using the Nikon Eclipse TS100 fluorescence microscope.
Differentiation Potential
Cryopreserved DMSCs were analyzed for their multilineage differentiation potential. Differentiation towards Osteogenic lineage was done as discussed previously. Briefly, 2 × 104 was plated in each well of 24-well plates. Osteogenic differentiation was induced by culturing cells for 21 days in the presence of complete α-MEM media supplemented with osteoinductive factors including dexamethasone (0.01 mM), β-glycerophosphate (5 mM), and monopotassium phosphate (1.8 mM) [26] obtained from Sigma. Differentiation media was replaced with fresh media every third day. At the end of the differentiation period, cells were stained by alizarin red stain (Sigma-Aldrich). Similarly, for adipogenic and chondrogenic differentiations, cells were plated at the seeding density of 2 × 104cells per 24-well plate and differentiations were induced by culturing in the presence of respective adipogenic and chondrogenic induction media (HiMedia) for 21 days. At the end of the incubation period, cells were stained with oil red O stain (Sigma-Aldrich) for assessment of adipogenic differentiation and cells induced with chondrogenic media were stained with alcian blue stain as per manufacturer’s instruction (HiMedia).
For neural differentiation, cells were cultured in the presence of neural basal media (Gibco, USA) supplemented with growth factors purchased from Invitrogen including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF) at the concentration of 20 ng/ml, and 1% B27 supplement for a period of 25 days [27]. Neurogenic differentiation potential of DMSC was assessed by immunostaining with neural-specific antibodies, i.e., anti-microtubule-associated protein (MAP-2) and anti-neurofilament antibodies (NFM). Briefly, cells were fixed and permeabilized followed by blocking of nonspecific antigen sites using bovine serum albumin (BSA) (Sigma-Aldrich). Cells were then incubated overnight at 4 °C with anti-human MAP-2 and NFM primary antibodies. After primary antibody incubation, cells were further incubated with FITC labeled anti-rabbit secondary antibody. Images were captured using the Nikon Eclipse TS100 fluorescence microscope. DAPI was used as a counterstain for the cell nucleus.
Karyotype Analysis
Briefly, DMSC cultures at their exponential phase of growth were arrested by giving KaryoMax colcemid (Gibco) treatment for 2 h at 37 °C. Cells were harvested by trypsinization and were subjected to hypotonic treatment with 0.075 M KCL followed by incubation at 37 °C for 15 min. Thereafter, cell fixation step was performed using methanol: glacial acetic acid (3:1) solution. For slide preparation, the cell suspension was dropwise made to fall on cold slides followed by drying at 40–42 °C for 1–2 min. Slides were subjected to methanol and trypsin treatment, and before staining, they were washed with cold water. The slide was stained with Giemsa stain (HiMedia) for an optimized period. The cells were then observed under the light microscope [28].
Results
Cryopreserved DMSC Culture Revival and Phenotype Characterization
Cryopreserved dental stem cell vials were successfully thawed after long-term storage at − 80 °C. After revival, 70% confluency of these DMSCs was observed within 7 days of the culture. Cells showed normal spindle-shaped morphology, typical of mesenchymal cells (Fig. 1a). Phenotypic characterization of these cryopreserved cells showed positive expression of CD90, CD73, and CD105 stemness markers and negative for CD34, CD45, and HLA-DR (Fig. 1b). The quantitative expression of the stemness marker for these cryopreserved stem cells was observed to be equivalent to freshly characterized DMSCs. The percentage positivity of fresh DMSCs was 91.8 ± 5.7, 95.4 ± 3, and 45.7 ± 11.4 for CD90, CD73, and CD105 respectively. Also, there was a similar expression of negative markers including endothelial, hematopoietic markers, and HLA-DR between cryopreserved and fresh DMSC (Fig. 1c).

Cryopreserved dental mesenchymal stromal cells morphology and characterization. a Confluent culture of cryopreserved DMSCs showing spindle-shaped morphology post-revival. b Dot plot showing positive expression of stemness markers (CD90, CD73, CD105) and negative expression of endothelial marker (CD34), hematopoietic markers (CD45), and HLA-DR in DMSCs after cryopreservation. c Bar diagram showing the positive percent of cells for different antibodies in fresh and cryopreserved samples. Magnification × 10. Scale bar 50 μm (n = 4).
Post-thaw Viability and Proliferation
Post-thaw proliferation potential of these cells was found to be unaffected after uncontrolled long-term storage as analyzed by colorimetric based EZblue assay (Fig. 2a). Also, the MTT assay revealed more than 95% of metabolically active cells (data not shown). The latter was further validated by Annexin-PI assay for total cell death (Fig. 2b); results indicated 84.2 ± 8.8% viable cell population as shown in Fig. 2c. Assessment of the cell growth status of cryopreserved DMSCs by determining the percentage distribution of cells at different phases of the cell cycle showed a similar trend as observed for cell proliferation results (Fig.2d). Percentage distribution of DMSCs was found to be 70.34 ± 4.4, 9.7 ± 1.7, and 19 ± 3.8 in three phases of cell cycle viz. Go/G1, S, and G2/M respectively and almost negligible for sub-G1 (Fig. 2e).

Effect of long-term cryopreservation on viability and proliferation ability of DMSCs. a Post-thaw DMSC proliferation analysis by EZBlue stain. b Dot plot image represents positive percentage of cells stained with Annexin/PI. c Bar diagram showing total percentage of live, apoptotic and necrotic cells. d Cell cycle phase distribution of cryopreserved stem cells in different stages of cell cycle. e Bar diagram showing cell percentage distribution of DMSC at different phase of cell cycle (n = 4)
Colony-Forming Units and Sphere Formation Ability
The colony-forming assay is another potential method for assessing the functionality of a stem cell that reveals the clonal expansion ability of the single-cell population. Colony-forming ability was found to be persistent in these DMSC as shown in Fig. 3a after uncontrolled long-term cryopreservation. The average number of colonies formed by these DMSC after cryopreservation was approx. 30 ± 17 (data not shown). Self-renewal ability was further assessed by subjecting these cells to grow under low adherent conditions. These cells were able to form three-dimensional spheroid when cultured in ultra-low attachment surface. The sphere formation started after 24 h (day 0), and by day 7, significant increase was observed in the size of the spheres with an average diameter of20.9 ± 4.7, 43.88 ± 7.7, 54.7 ± 8.5, and 61.71 ± 10.3 μm at days 0, 3, 5 and 7 respectively (Fig. 3b, c). We also assessed the viability of these spheroids by staining them with AO-EtBr dual stain. The results indicated viable spheroids at day 7 as shown in Fig. 3d.

Representative images of colony-forming assay and spheres forming ability of DMSCs after long-term cryopreservation. a Colony-formation in cryopreserved DMSCs after 7 days of culture, stained with crystal violet. Inset picture represents an individual well image of a six-well plate. b Brightfield images showing spheres formation from day 0 (day of plating), day 3, day 5, and day 7. c Bar diagram represents the size of the spheres increase from day 0 to day 7. d Images of viable spheres with intact membrane integrity at day 7 stained with AO/EtBr stain. Magnification × 4, Scale bar 50 μm
Differentiation Potential of Cryopreserved DMSCs
Furthermore, the multipotency of these cryopreserved DMSCs was assessed by inducing them to differentiate into osteogenic, adipogenic, chondrogenic, and neural lineages. As depicted in Fig. 4a, we observed mineral nodules in osteogenically induced cryo-DMSCs when stained with alizarin red stain similar to fresh DMSCs, indicating that these cells after cryopreservation retained their osteogenic potential on receiving osteogenic stimulation for 21 days. Similarly, cryopreserved DMSCs induced to adipogenic lineage stained positively for oil red O stain, which confirmed the formation of lipid droplets in these induced cells, hence their differentiation ability towards adipogenic lineage (Fig. 4a).

Post-thaw differentiation ability of cryopreserved DMSC. a Alizarin red stained mineral nodules (upper panel), Oil red O–stained lipid droplets (middle) and alcian blue staining (lower) images show differentiated DMSC after giving stimulation towards the respective lineage in fresh and cryopreserved DMSCs. b Immunostaining images (neurofilament and microtubule associated protein) showing neural potential of cryopreserved DMSCs after long-term cryopreservation. Magnification × 10 and × 20, Scale bar 50 μm
Next, we assessed the chondrogenic differentiation ability of cryo-DMSCs after the induction period. These cryopreserved DMSCs showed positivity for alcian blue stain as indicated in Fig. 4a.
Also, the neurogenic potential of these cryo-DMSC cells was determined by immunostaining with anti-map and NFM antibodies that are specific for neurogenic lineage. Immunostaining results revealed the positivity of these cells for neural-specific protein viz. MAP and NFM (Fig. 4b).
Overall, multilineage differentiation results suggested that long-term uncontrolled cryopreservation does not impede the multilineage potential of these dental mesenchymal stem cells.
Karyotyping
At the cytogenetic level, the stability of DMSCs after long-term storage at − 80 °C followed by post-revival culture expansion was determined by performing karyotype. No numerical or structural abnormalities were observed in the karyotype as shown in Fig. 5.

Karyotype of cryopreserved DMSC: representative image of karyotype (G-banded) of DMSCs stored for up to 5 years
Discussion
The demand for cell-based therapies is slowly gaining importance in regenerative medicine. Stem cells, with their unique self-renewing ability, render an unlimited pool of cells that can be cryopreserved for future clinical and research applications. Although, the emergence of modern cryopreservation technologies facilitates stem cell banking for subsequent administration which has resolved issues like donor availability and donor attrition [29]. But the successful clinical implementation of stem cell-based therapy relies upon constructive and robust cryopreservation approaches, which can efficiently achieve a consistent outcome in terms of maximal viable cell recovery, functionality, and immunomodulation after prolonged storage [19, 30].
The conventional cryopreservation method includes slow cooling in the presence of a cryoprotectant usually DMSO in combination with the fetal bovine serum that protects the cells from rapid freezing and thawing cycles [12]. DMSO can penetrate the cells, decrease the water incorporation into the cells, and protect the cells from excessive dehydration. Ten percent of concentration of DMSO is optimized for the preservation of cells and often recommended to avoid the adverse reactions related to the infusion of DMSO [31]. The controlled rate freezing technique at 1 to 2 °C/min and rapid thawing are considered standard and are routinely followed. However, in our study, we observed that the DMSCs could be successfully cryopreserved for a long time by the uncontrolled freezing method.
In a previous study, we were able to recover the stem cells which were stored by uncontrolled freezing in 10% DMSO at − 80 °C for at least 1 year. The dental pulp stem cells (DPSCs) retained their biological as well as functional properties even after 1 year of uncontrolled freezing [24]. In the present study, we endeavored to study the post-thaw recovery of dental mesenchymal stem cell samples (DPSC, DFSC) which have been stored at − 80 °C for up to 5 years. After thawing, these cells adhered to the plastic surface and achieved 70% confluence within,7 days. Direct cryopreservation of cells is known to induce alteration in the structure and binding properties of the cell membrane. However, previously our group along with others have reported the impact of different concentrations of cryopreservation agents and cryopreservation methods on revival and survival of stem cells from dental tissue and found 10% DMSO as an optimum concentration for preserving and recovering viable DMSC without major loss of functionality at − 80 °C [24, 32]. Further, we also observed more than 90% expression for mesenchymal stem cell markers like CD90, CD73, and CD105 and negative expression of CD34, CD45, and HLA-DR in these cells which imply that dental mesenchymal stem cells can survive long-term uncontrolled freezing without losing their phenotypic characteristics.
DMSO, which is added as a cryoprotectant for long-term storage, is known to be cell-permeable and interacts with cell metabolism leading to cell toxicity, disrupting the cell cytoskeleton and eventually death [33, 34]. However, we observed that cryopreservation using 10% DMSO had no detrimental effect on viability, proliferation, or cell cycle pattern of DMSCs. A few studies have reported the adverse effects of DMSO on cell proliferation, e.g. Fiore et al. found that DMSO induced cell arrest at G1 phase of the cell cycle in Chinese hamster cells (CHO cells) [35,36,37,38], but in our case, the results of EZblue and MTT assays showed that these cells were metabolically active with persistent proliferative abilities even after 5 years of uncontrolled cryopreserved conditions. Recently, Kumar et al. have reported that cell viability in human adipose–derived stem cells (hASCs) after decade long cryopreservation depends on many factors including patient age, passage number, and also the methods of cryopreservation/thawing adopted by different labs across the world [39]. Cell viability data were validated with annexin-PI based cell apoptosis assay, where more than 85% of cell viability was observed in post-thaw samples. Further, results of cell cycle analysis were found to be consistent with our previous findings, indicating that DMSO has no inhibitory effect on cell proliferation of cryopreserved DMSCs even after long-term cryopreservation up to 5 years. Thus, dental mesenchymal stromal cells can resist harsh conditions without compromising cell integrity and can serve as a potent source of stem cells to be used in cell banking for future clinical implementations. One of the potential reasons attributed to cell attachment, viability, and unaltered proliferation abilities included the cryoprotective nature of serum in cryomedia. The addition of FBS has shown to increase the efficiency of cryopreservation, unlike its complete elimination which drastically reduced the survival of stem cells including mesenchymal stromal cells and embryonic stem cells [40, 41]. Furthermore, a higher concentration of FBS up to 90% in cryomedia had more potential in reducing freezing and thawing cycles associated with damage to the eukaryotic cell as compared to lower FBS concentration [42, 43].
The colony-forming assay is a gold standard method to assess the property of stem cells including self-renewal ability, cellular proliferation, and differentiation capacity [25]. In a previous study, an inverse relationship was reported between DMSO concentration and colony-forming units. The authors observed that cryopreservation using 20%DMSO significantly hampered the colony-forming abilities of these cells [44]. Similarly, decreased cell viability, expression of adhesion molecule α4 integrin, and colony-forming ability were observed for hASCs cryopreserved in 10% DMSO [45]. However, in our case, the colony-forming ability of cryopreserved DMSC remain unaffected at the same concentration of DMSO, thus suggesting that cryopreservation does not compromise the biological properties of dental mesenchymal stromal cells, unlike various other stem cells that are more susceptible to loss of function on long-term storage [46].
The self-renewal ability of stem cells at a single cell level is also facilitated by the sphere-forming assay [47] which can take place by growing cells at non- adherent conditions. This assay allowed us to examine the sphere-forming ability after prolonged cryopreservation, which was found to be retained in these stem cells. Moreover, most of the spheres were found to be viable on day7. These results further validated the fact that dental mesenchymal stromal cell’s biological properties are not impeded by long-term preservation at − 80 °C and can be stored for future implications in cost-effective ways.
Clinical implications of stem cells are attributed to their multilineage differentiation potential. Dental mesenchymal cells can differentiate into mesoderm, endoderm, as well as ectoderm lineages [48, 49]. This potential of dental stem cells opens the door for various therapeutic applications and cell banking. However, reduced differentiation ability resulted from cryopreservation has been reported by many groups [50]. In our study, we observed the intact differentiation ability of dental mesenchymal stem cells after uncontrolled cryopreservation for 5 years. These cells were able to differentiate into osteogenic lineage after induction for 21 days. Similarly, positive staining of fat globules within the differentiated stem cells by oil red O staining also suggested the persistence of adipogenic potential. Our results also confirmed the chondrogenic differentiation potential after cryopreservation. Being neuroectodermal in origin, these dental mesenchymal stromal cells can differentiate into nerve cells upon stimulation [51]. We observed positivity for neural differentiation biomarkers like microtubule-associated proteins as well as neurofilament proteins. Besides the cryopreservation technique and procedure, other possible reasons for the retained biological properties such as proliferation, colony formation, and differentiation by these long-term uncontrolled cryopreserved DMSCs might be associated with the donor’s age. In our study, the inclusion criteria opted for the collection and subsequently, cryopreservation of DMSCs included individuals with or below 25 years of age. Several reports have shown that the biological properties and functionality of stem cells are age-dependent, and with an increase in donor age, there is a deterioration in the properties of mesenchymal stromal stem [52, 53]. Further, no signs of chromosomal instability were observed following scale-up of the process of these cryopreserved DMSCs, thus confirming the genetic stability of these preserved stem cells.
Conclusion
Thus, it can be concluded that stem cells from dental origin are capable of withstanding the harsh conditions and can be cryopreserved cost-effectively without altering their biological properties. The plastic-adherent DMSCs can be expanded in culture after long-term uncontrolled cryopreservation without the loss of their multipotency. This study encourages to investigate the ability of these cryopreserved dental mesenchymal stromal cells in various in vivo diseased models, which is an important translational aspect of stem cell research. This would reinforce the idea of present work, where we consider dental mesenchymal stem as a potent source of stem cells for future cell banking as well as clinical applications as illustrated in the graphical abstract.
Future Perspective
Future insights, as well as in vivo investigations, would be beneficial to confirm their potent role in various disease conditions. Nevertheless, we provide very strong evidence for considering these dental mesenchymal stromal cells as an ideal source to be utilized clinically as well as in cryobanking.
References
Schallmoser, K., Rohde, E., Reinisch, A., Bartmann, C., Thaler, D., Drexler, C., Obenauf, A. C., Lanzer, G., Linkesch, W., & Strunk, D. (2008). Rapid large-scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Engineering Part C, Methods, 14(3), 185–196. https://doi.org/10.1089/ten.tec.2008.0060.
Mckee, C., & Chaudhry, G. R. (2017). Advances and challenges in stem cell culture. Colloids and Surfaces B Biointerfaces, 159, 62–77. https://doi.org/10.1016/j.colsurfb.2017.07.051.
Kropp, C., Massai, D., & Zweigerdt, R. (2017). Progress and challenges in large-scale expansion of human pluripotent stem cells. Process Biochemistry, 59, 244–254. https://doi.org/10.1016/j.procbio.2016.09.032.
Ratcliffe, E., Thomas, R. J., & Williams, D. J. (2011). Current understanding and challenges in bioprocessing of stem cell-based therapies for regenerative medicine. British Medical Bulletin, 100(1), 137–155. https://doi.org/10.1093/bmb/ldr037.
Turinetto, V., Vitale, E., & Giachino, C. (2016). Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. International Journal of Molecular Sciences, 17(7), 1164. https://doi.org/10.3390/ijms17071164.
Safwani, W. K. Z. W., Makpol, S., Sathapan, S., & Chua, K. H. (2012). The impact of long-term in vitro expansion on the senescence-associated markers of human adipose-derived stem cells. Applied Biochemistry and Biotechnology, 166(8), 2101–2113. https://doi.org/10.1007/s12010-012-9637-4.
Pal, R., Hanwate, M., & Totey, S. M. (2008). Effect of holding time, temperature and different parenteral solutions on viability and functionality of adult bone marrow-derived mesenchymal stem cells before transplantation. Journal of Tissue Engineering and Regenerative Medicine, 2(7), 436–444. https://doi.org/10.1002/term.109.
Di Wu, Y., Li, M., Liao, X., Li, S. H., Yan, J. X., Fan, L., et al. (2019). Effects of storage culture media, temperature and duration on human adipose-derived stem cell viability for clinical use. Molecular Medicine Reports, 19(3), 2189–2201. https://doi.org/10.3892/mmr.2019.9842.
Park, B.-W., Jang, S.-J., Byun, J.-H., Kang, Y.-H., Choi, M.-J., Park, W.-U., Lee, W. J., & Rho, G.-J. (2017). Cryopreservation of human dental follicle tissue for use as a resource of autologous mesenchymal stem cells. Journal of Tissue Engineering and Regenerative Medicine, 11(2), 489–500. https://doi.org/10.1002/term.1945.
Yang, H., Li, J., Sun, J., Guo, W., Li, H., Chen, J., Hu, Y., Tian, W., & Li, S. (2017). Cells isolated from cryopreserved dental follicle display similar characteristics to cryopreserved dental follicle cells. Cryobiology, 78, 47–55. https://doi.org/10.1016/j.cryobiol.2017.07.003.
Li, Y., Ph, D., Tan, J., Ph, D., Li, L., & Ph, D. (2010). Comparison of three methods for cryopreservation of human embryonic stem cells. Fertility and Sterility, 93(3), 999–1005. https://doi.org/10.1016/j.fertnstert.2008.10.052.
Duan, W., Lopez, M. J., & Hicok, K. (2018). Adult multipotent stromal cell cryopreservation: pluses and pitfalls. Veterinary Surgery, 47(1), 19–29. https://doi.org/10.1111/vsu.12730.
Haddock, R., Lin-Gibson, S., Lumelsky, N., McFarland, R., Roy, K., Saha, K., et al. (2017). Manufacturing cell therapies: the paradigm shift in health care of this century. NAM Perspectives, 7(6), 1–13. https://doi.org/10.31478/201706c.
Gao, D., & Critser, J. K. (2000). Mechanisms of cryoinjury in living cells. ILAR Journal, 41(4), 187–196. https://doi.org/10.1093/ilar.41.4.187.
Li, Y., & Ma, T. (2012). Bioprocessing of cryopreservation for large-scale banking of human pluripotent stem cells. BioResearch Open Access, 1(5), 205–214. https://doi.org/10.1089/biores.2012.0224.
Hoon, T., Choel, S., Hyun, J., Yoon, J., Hong, J., Seo, U., et al. (2017). Cryopreservation and its clinical applications. Integrative Medicine Research, 6(1), 12–18. https://doi.org/10.1016/j.imr.2016.12.001.
Doğan, A., Yalvaç, M. E., Yılmaz, A., Rizvanov, A., & Şahin, F. (2013). Effect of f68 on cryopreservation of mesenchymal stem cells derived from human tooth germ. Applied Biochemistry and Biotechnology, 171(7), 1819–1831. https://doi.org/10.1007/s12010-013-0472-z.
Baboo, J., Kilbride, P., Delahaye, M., Milne, S., Fonseca, F., Blanco, M., Meneghel, J., Nancekievill, A., Gaddum, N., & Morris, G. J. (2019). The impact of varying cooling and thawing rates on the quality of cryopreserved human peripheral blood T cells. Scientific Reports, 9(1), 1–13. https://doi.org/10.1038/s41598-019-39957-x.
Hunt, C. J. (2011). Cryopreservation of human stem cells for clinical application: a review. Transfusion Medicine and Hemotherapy, 38(2), 107–123. https://doi.org/10.1159/000326623.
Dricu, A., & Dricu, A. (2018). Expert opinion on biological therapy recent challenges with stem cell banking recent challenges with stem cell banking. Expert Opinion on Biological Therapy, 18(4), 355–358. https://doi.org/10.1080/14712598.2018.1445715.
Yuan, Z., Da, S., Lourenco, S., Sage, E. K., Kolluri, K. K., Lowdell, M. W., & Janes, S. A. M. M. (2016). Cryopreservation of human mesenchymal stromal cells expressing TRAIL for human anti-cancer therapy. Cytotherapy, 18(7), 860–869. https://doi.org/10.1016/j.jcyt.2016.04.005.
Le Blanc, K., & Davies, L. C. (2018). MSCs—cells with many sides. Cytotherapy, 20(3), 273–278. https://doi.org/10.1016/j.jcyt.2018.01.009.
Yuan, Y., Yang, Y., Tian, Y., Park, J., Dai, A., Roberts, R. M., Liu, Y., & Han, X. (2016). Efficient long-term cryopreservation of pluripotent stem cells at −80 °C. Scientific Reports, 6(1), 34476. https://doi.org/10.1038/srep34476.
Kumar, A., Bhattacharyya, S., & Rattan, V. (2015). Effect of uncontrolled freezing on biological characteristics of human dental pulp stem cells. Cell and Tissue Banking, 16(4), 513–522. https://doi.org/10.1007/s10561-015-9498-5.
Digirolamo, C. M., Stokes, D., Colter, D., Phinney, D. G., Class, R., & Prockop, D. J. (1999). Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. British Journal of Haematology, 107(2), 275–281. https://doi.org/10.1046/j.1365-2141.1999.01715.x.
Bakopoulou, A., Leyhausen, G., Volk, J., Tsiftsoglou, A., Garefis, P., Koidis, P., & Geurtsen, W. (2011). Comparative analysis of in vitro osteo/odontogenic differentiation potential of human dental pulp stem cells (DPSCs) and stem cells from the apical papilla (SCAP). Archives of Oral Biology, 56(7), 709–721. https://doi.org/10.1016/j.archoralbio.2010.12.008.
Kumar, A., Kumar, V., Rattan, V., Jha, V., & Bhattacharyya, S. (2017). Secretome cues modulate the neurogenic potential of bone marrow and dental stem cells. Molecular Neurobiology, 54(6), 4672–4682. https://doi.org/10.1007/s12035-016-0011-3.
Borgonovo, T., Vaz, I. M., Senegaglia, A. C., Rebelatto, C. L. K., & Brofman, P. R. S. (2014). Genetic evaluation of mesenchymal stem cells by G-banded karyotyping in a cell technology center. Revista Brasileira de Hematologia e Hemoterapia, 36(3), 202–207. https://doi.org/10.1016/j.bjhh.2014.03.006.
Harris, D. (2016). Long-term frozen storage of stem cells: challenges and solutions. Journal of Biorepository Science for Applied Medicine, 4, 9–20. https://doi.org/10.2147/bsam.s90142.
Hanna, J., & Hubel, A. (2009). Preservation of stem cells. Organogenesis, 5(3), 134–137. https://doi.org/10.4161/org.5.3.9585.
CAVINS, J. A., KASAKURA, S., THOMAS, E. D., & FERREBEE, J. W. (1963). Recovery of lethally irradiated dogs following infusion of autologous marrow stored at low temperature in dimethyl-sulfoxide. JAMA, 183(1), 179. https://doi.org/10.1001/jama.1963.03700010133077.
Woods, E. J., Perry, B. C., Hockema, J. J., Larson, L., Zhou, D., & Goebel, W. S. (2009). Optimized cryopreservation method for human dental pulp-derived stem cells and their tissues of origin for banking and clinical use. Cryobiology, 59(2), 150–157. https://doi.org/10.1016/j.cryobiol.2009.06.005.
Naaldijk, Y., Staude, M., Fedorova, V., & Stolzing, A. (2012). Effect of different freezing rates during cryopreservation of rat mesenchymal stem cells using combinations of hydroxyethyl starch and dimethylsulfoxide. BMC Biotechnology, 12(1), 49. https://doi.org/10.1186/1472-6750-12-49.
Penninckx, F., Cheng, N., Kerremans, R., Van Damme, B., & de Loecker, W. (1983). The effects of different concentrations of glycerol and dimethylsulfoxide on the metabolic activities of kidney slices. Cryobiology, 20(1), 51–60. https://doi.org/10.1016/0011-2240(83)90059-7.
De Abreu Costa, L., Ottoni, M. H. F., Dos Santos, M. G., Meireles, A. B., De Almeida, V. G., De Fátima Pereira, W., et al. (2017). Dimethyl sulfoxide (DMSO) decreases cell proliferation and TNF-α, IFN-, and IL-2 cytokines production in cultures of peripheral blood lymphocytes. Molecules, 22(11), 1–10. https://doi.org/10.3390/molecules22111789.
Jamalzadeh, L., Ghafoori, H., Sariri, R., Rabuti, H., Nasirzade, J., Hasani, H., & Aghamaali, M. R. (2016). Cytotoxic effects of some common organic solvents on MCF-7, RAW-264.7 and human umbilical vein endothelial cells. Avicenna Journal of Medical Biochemistry, In press(In press), 10–33453. https://doi.org/10.17795/ajmb-33453.
Öz, E. S., Aydemir, E., & Fişkin, K. (2012). DMSO exhibits similar cytotoxicity effects to thalidomide in mouse breast cancer cells. Oncology Letters, 3(4), 927–929. https://doi.org/10.3892/ol.2012.559.
Fiore, M., Zanier, R., & Degrassi, F. (2002). Reversible G(1) arrest by dimethyl sulfoxide as a new method to synchronize Chinese hamster cells. Mutagenesis, 17(5), 419–424. https://doi.org/10.1093/mutage/17.5.419.
Kumar, A., Xu, Y., Yang, E., Wang, Y., & Du, Y. (2019). Fidelity of long-term cryopreserved adipose-derived stem cells for differentiation into cells of ocular and other lineages. Experimental Eye Research, 189, 107860. https://doi.org/10.1016/j.exer.2019.107860.
Shivakumar, S. B., Bharti, D., Jang, S.-J., Hwang, S.-C., Park, J.-K., Shin, J.-K., et al. (2015). Cryopreservation of human Wharton’s jelly-derived mesenchymal stem cells following controlled rate freezing protocol using different cryoprotectants; a comparative study. International Journal of Stem Cells, 8(2), 155–169. https://doi.org/10.15283/ijsc.2015.8.2.155.
Ha, S. Y., Jee, B. C., Suh, C. S., Kim, H. S., Oh, S. K., Kim, S. H., & Moon, S. Y. (2005). Cryopreservation of human embryonic stem cells without the use of a programmable freezer. Human Reproduction, 20(7), 1779–1785. https://doi.org/10.1093/humrep/deh854.
Renzi, S., Lombardo, T., Dotti, S., Dessì, S. S., De Blasio, P., & Ferrari, M. (2012). Mesenchymal stromal cell cryopreservation. Biopreservation and Biobanking, 10(3), 276–281. https://doi.org/10.1089/bio.2012.0005.
Cristian, M., Conde, M., Chisini, L. A., Grazioli, G., Francia, A., De Carvalho, R. V., et al. (2016). Does cryopreservation affect the biological properties of stem cells from dental tissues? A systematic review. Brazilian Dental Journal, 27(6), 633–640. https://doi.org/10.1590/0103-6440201600980.
Ock, S. A., & Rho, G. J. (2011). Effect of dimethyl sulfoxide (DMSO) on cryopreservation of porcine mesenchymal stem cells (pMSCS). Cell Transplantation, 20(8), 1231–1239. https://doi.org/10.3727/096368910X552835.
Irioda, A. C., Cassilha, R., Zocche, L., Francisco, J. C., Cunha, R. C., Ferreira, P. E., et al. (2016). Human adipose-derived mesenchymal stem cells cryopreservation and thawing decrease α 4-integrin expression. Stem Cells International, 2016, 1–9. https://doi.org/10.1155/2016/2562718.
James, A. W., Levi, B., Nelson, E. R., Peng, M., Commons, G. W., Lee, M., Wu, B., & Longaker, M. T. (2011). Deleterious effects of freezing on osteogenic differentiation of human adipose-derived stromal cells in vitro and in vivo. Stem Cells and Development, 20(3), 427–439. https://doi.org/10.1089/scd.2010.0082.
Pastrana, E., Silva-Vargas, V., & Doetsch, F. (2011). Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell, 8(5), 486–498. https://doi.org/10.1016/j.stem.2011.04.007.
Gronthos, S., Brahim, J., Li, W., Fisher, L. W., Cherman, N., Boyde, A., DenBesten, P., Robey, P. G., & Shi, S. (2002). Stem cell properties of human dental pulp stem cells. Journal of Dental Research, 81(8), 531–535. https://doi.org/10.1177/154405910208100806.
itkowska-Zimny, M. W. (2012). Dental tissue as a source of stem cells: perspectives for teeth regeneration. Journal of Bioengineering and Biomedical Sciences, 01(S2), 2–5. https://doi.org/10.4172/2155-9538.s2-006.
Qi, W., Ding, D., & Salvi, R. J. (2008). Cytotoxic effects of dimethyl sulphoxide (DMSO) on cochlear organotypic cultures. Hearing Research, 236(1–2), 52–60. https://doi.org/10.1016/j.heares.2007.12.002.
Potdar, P. D. (2015). Human dental pulp stem cells: applications in future regenerative medicine. World Journal of Stem Cells, 7(5), 839–851. https://doi.org/10.4252/wjsc.v7.i5.839.
Horibe, H., Murakami, M., Iohara, K., Hayashi, Y., Takeuchi, N., Takei, Y., Kurita, K., & Nakashima, M. (2014). Isolation of a stable subpopulation of mobilized dental pulp stem cells (MDPSCs) with high proliferation, migration, and regeneration potential is independent of age. PLoS One, 9(5), e98553. https://doi.org/10.1371/journal.pone.0098553.
Volk, S. W., Wang, Y., & Hankenson, K. D. (2012). Effects of donor characteristics and ex vivo expansion on canine mesenchymal stem cell properties: implications for MSC-based therapies. Cell Transplantation, 21(10), 2189–2200. https://doi.org/10.3727/096368912X636821.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Written consent was signed by the patient, and all the protocols were approved by the Institute Ethics Committee.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Raik, S., Kumar, A., Rattan, V. et al. Assessment of Post-thaw Quality of Dental Mesenchymal Stromal Cells After Long-Term Cryopreservation by Uncontrolled Freezing. Appl Biochem Biotechnol 191, 728–743 (2020). https://doi.org/10.1007/s12010-019-03216-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-019-03216-6