
Abstract
A heterologous xylose utilization pathway, either xylose reductase–xylitol dehydrogenase (XR–XDH) or xylose isomerase (XI), is usually introduced into Saccharomyces cerevisiae to construct a xylose-fermenting strain for lignocellulosic ethanol production. To investigate the molecular basis underlying the effect of different xylose utilization pathways on the xylose metabolism and ethanol fermentation, transcriptomes of flocculating industrial strains with the same genetic background harboring different xylose utilization pathways were studied. A different source of xylA did not obviously affect the change of the strains transcriptome, but compared with the XR–XDH strain, several key genes in the central carbon pathway were downregulated in the XI strains, suggesting a lower carbon flow to ethanol. The carbon starvation caused by lower xylose metabolism in XI strains further influenced the stress response and cell metabolism of amino acid, nucleobase, and vitamin. Besides, the downregulated genes mostly included those involved in mitotic cell cycle and the cell division–related process. Moreover, the transcriptomes analysis indicated that the after integrate xylA in the δ region, the DNA and chromosome stability and cell wall integrity of the strains were affected to some extent. The aim of this was to provide some reference for constructing efficient xylose-fermenting strains.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass is a renewable raw material for bioethanol production to avoid a food crisis caused by grain ethanol production [1]. Saccharomyces cerevisiae is extensively used for bioethanol production because of its excellent environmental tolerance and high ethanol fermentation productivity [2]. However, native S. cerevisiae was almost impossible to utilize xylose [3], in which the glucose content was only lower than in lignocellulosic hydrolysate. It is of economic interest to engineer industrial S. cerevisiae strains with xylose metabolic capacity [4]. To achieve this objective, one of the two heterologous xylose catabolism pathways, the xylose reductase–xylitol dehydrogenase (XR–XDH) pathway or xylose isomerase (XI) pathway, is usually introduced into S. cerevisiae [2].
The main challenge with the strains harboring the recombinant XR–XDH pathway is the redox imbalance because the reduction of xylose to xylitol was catalyzed by XR preferably using NADPH, but the oxidation of xylitol to xylulose was catalyzed by XDH strictly using NAD+, leading to the accumulation of the xylitol and reduced ethanol yields. On the other hand, no intermediate product is generated in the XI pathway as xylose is directly metabolized to xylulose without a coenzyme by XI. However, the xylose consumption rate of XI strains is usually slower than that of XR–XDH strains [5].
Besides the metabolism of xylose to xylulose per se, the expression of different heterologous xylose utilization pathways may have an overall effect on the metabolic process resulting in different xylose consumption rates and ethanol yields. Identification of the molecular mechanism underlying the effect of different xylose utilization pathways is of interest in basic and applied research for bioethanol production. Metabolomics studies indicate that the XI strains exhibit a stronger carbon starvation response than the XR–XDH strain [6]. However, from our knowledge, there is no study reporting the transcriptional response differentiation between the XR–XDH and XI pathways.
In previous studies, we introduced different heterologous xylose utilization pathways in the same host strain. The flocculating industrial S. cerevisiae strain NAPX37, harboring the XR–XDH pathway, has superior fermentation capability and inhibitor tolerance [7]. Strain Alpha25 is the haploid of NAPX37. The fragment XYL1–XYL2 was deleted from Alpha25, resulting in strain YC-8 [7]. The codon-optimized xylA from Orpinomyces sp. or Prevotella ruminicola was then introduced into the δ region of the genome of strain YC-8, generating strains O7 and P5, respectively. In this study, a comparative transcriptional analysis was performed using RNA-Seq to unravel the global gene expression difference induced by different xylose utilization pathways. The isogenic relationship among the strains ascertained that the observed differences were because of the particular xylose utilization pathway and not due to unknown differences in regulatory systems. The purpose of this research was to provide some reference for constructing efficient xylose-fermenting strains.
Materials and Methods
Strain Construction
All the S. cerevisiae strains used in this study were listed in Supplementary Materials 1: Table S1. NAPX37 harbors the XR–XDH pathway [7]. The haploid strain, Alpha25, was germinated from NAPX37 in the previous study [8]. The XR–XDH pathway of Alpha25 was knocked out by homologous recombination to generate strain YC-8 [8]. The codon-optimized xylA from Orpinomyces sp. (OrxylA2; accession number KY630521) and P. ruminicola (PrxylA2; accession number KY630522) were separately multi-expressed into the yeast genome by δ-integration. The fragments PTHD3–OrxylA2–TTDH3 and PTHD3–PrxylA2–TTDH3 were amplified from the plasmids pRS426-OXYLA2 and pRS426-PXYLA2 [8], respectively, using the primers δ-XI-F (tgttggaatagaaatcaactatcatctactaactggagtgatgcaacctgcctggag) and δ-XI-R (gtttatattcattgatcctattacattacgccagggttttcccagtcacgac) that harbored the sequences for homologous recombination. The fragments were δ-integrated into the host strain YC-8. The transformants O7 and P5 with the best xylose fermentation capacities and similar xylA copies, one with OrxylA2 and the other with PrxylA2, respectively, were selected [9].
Xylose Fermentation
Yeast strains were cultured under aerobic conditions at 30 °C in 5% YPD medium (peptone 20 g/L, yeast extract 10 g/L, glucose 50 g/L) for 48 h. Cells were collected by centrifugation at 8000g for 2 min. The cell pellets were washed twice with distilled water and inoculated to 4% YPX medium (peptone 20 g/L, yeast extract 10 g/L, and xylose 40 g/L), 10% YPDX medium (peptone 20 g/L, yeast extract 10 g/L, xylose 40 g/L, and glucose 60 g/L), or lignocellulosic hydrolysate for fermentation. The initial cell concentration was adjusted to 0.23 g dry cell weight (DCW). The fermentation temperature was 35 °C and agitation speed was 120 rpm. For the lignocellulosic hydrolysate, bagasse was pretreated using the diluted H2SO4 method, as previously described [10], with minor modifications: 50 g of 0.5% (w/v) H2SO4 was mixed with 50 g dry bagasse. The hydrolysate was adjusted to pH 5, xylose 40 g/L, and glucose 60 g/L before being used for fermentation.
Sugar concentrations were measured by high-performance liquid chromatography (HPLC, LC-10AD VP, Shimadzu, Japan) established previously [11]. Ethanol concentrations were determined by gas chromatography (GC 353B, GL Sciences, Japan) according to the method described by Tang et al. [11]. Glycerol and xylitol were analyzed by HPLC (SCL-10A VP, Shimadzu, Japan) method developed previously [7].
RNA Sequencing and Transcriptome Analysis
Total RNA was extracted at 24 h during the fermentation with xylose as the sole carbon source according to the method described previously [9]: RNA concentration was measured using The Qubit®RNA Assay Kit in Qubit® 2.0 Fluorimeter (Life Technologies, CA, USA). RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). The RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Each RNA sample used for sequencing was the mixture of two biological replicates.
RNA-Seq libraries were constructed and sequenced at Sangon Biotech (Shanghai, China) by using Illumina Hiseq 2500 platform according to the method described by Gao et al. [12]. The raw sequence data of Alpha25, O7, and P5 can be obtained through the NCBI accession numbers SRR5251644, SRR5251643, and SRR5251640, respectively. The comparative transcriptome was analyzed according to the procedures described previously [9].
Results and Discussion
Physiology Differences of Xylose Metabolism in the XR–XDH and XI Strains
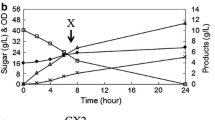
Two kinds of xylose utilization pathways, XR–XDH and XI, were separately expressed in the same strain, resulting in the strain Alpha25 that harbored Scheffersomyces stipitis XYL1–XYL2, strain O7 expressed codon-optimized OrxylA2, and strain P5 expressed codon-optimized PrxylA2. To compare their xylose utilization capabilities, batch fermentation was performed using 4% YPX with an initial OD660 of 0.8. The XR–XDH strain Alpha25 utilized xylose to proliferate and produced ethanol rapidly from the beginning of the fermentation (Fig. 1). In the first 8-h fermentation, 13.84 g/L xylose was consumed. Meanwhile, the XI strains O7 and P5 grew slowly in the first 8-h fermentation, and consumed 4.27 g/L xylose and 3.48 g/L xylose, respectively (Fig. 1b, c). After 48-h fermentation, 35.94 g/L xylose was consumed by Alpha25, whereas O7 and P5 consumed 17.71 g/L xylose and 26.10 g/L xylose, respectively (Table 1). The xylose consumption rate of Alpha25 was 103% higher than that of O7 and 38% higher than that of P5. However, Alpha25 accumulated 11.21 g/L of the byproduct xylitol, which corresponds to 31.2% of the consumed xylose. The XI strains O7 and P5 did not produce xylitol, and the ethanol yield reached at 0.396 g/g-xylose and 0.400 g/g-xylose, respectively (Table 1), which was 26.9% and 28.2% higher than that of Alpha25. The efficiencies of conversion from xylose to ethanol were 77.6% and 78.4% for O7 and P5, respectively.

Fermentation profiles of the recombinant strains with different xylose utilization pathways in medium containing xylose as the sole carbon source. a Alpha25. b O7. c P5. (Circle, OD660; square, xylose; diamond, ethanol; triangle, glycerol; error mark, xylitol)
In the glucose–xylose co-fermentation (Supplementary Materials 1: Fig. S1), glucose was completely consumed in the first 8-h fermentation for both the XI strains and XR–XDH strain, and they all proliferated rapidly. Meanwhile, Alpha25 consumed 8.97 g/L of xylose in the first 8-h fermentation, which was 2.02 times more than that of O7 and 3.54 times more than that of P5. After 48-h fermentation, Alpha25 consumed nearly all the xylose available (36.57 g/L), and the ethanol yield based on consumed sugar was 0.339 g/g. Although O7 and P5 consumed 46.8% and 47.0% less xylose than Alpha25, respectively, they did not accumulate xylitol, and the ethanol yields based on the consumed sugar were as high as 0.432 and 0.411 g/L, respectively (Supplementary Materials 1: Fig. S1). The results of the fermentation of lignocellulosic hydrolysate (Supplementary Materials 1: Fig. S2) indicated the same characteristics that Alpha25 exhibited stronger xylose utilization capability than O7 and P5; however, because of the accumulation of xylitol, its ethanol yield was lower than the XI strains.
Overview of the Transcriptional Differences in the XR–XDH and XI Strains
The effect of different source of xylA (O7 and P5) on the transcriptome was shown in Supplementary Materials 2. There were only 9 and 17 genes significantly upregulated and downregulated for O7 vs P5, respectively. So we thought a different source of xylA did not obviously affect the change of the strains transcriptome. But from the gene expression profiles shown in Supplementary Materials 2, it is indicated that the expression of different xylose utilization pathways (XI vs XR–XDH) disturbs the expression of the gene expressions. Therefore, in the present study, we focused on the transcriptome differences of strains with different xylose utilization pathways.
Although the genetic backgrounds of these strains were the same, the transcriptional profiles exhibited a distinct difference among the O7, P5, and Alpha25 strains (Supplementary Materials 2). Compared with Alpha25, 241 genes in O7 were expressed significantly different (log2(fold change) ≥ 1), among them, 144 upregulated and 97 downregulated, whereas 377 genes in P5 were expressed significantly different (log2(fold change) ≥ 1), among them, 188 upregulated and 189 downregulated. The number of genes that were significantly upregulated in both O7 and P5 was 83, and significantly downregulated in both O7 and P5 was 85 (Supplementary Materials 2).
The genes with significantly different expression levels in both O7 and P5 compared with Alpha25 were considered to be the conserved regulation module between the XI and XR–XDH pathways. The GO analysis identified that the observed expression differences mostly involved the transmembrane transport (GO:0055085), cellular amino acid metabolic process (GO:0006520), and mitotic cell cycle (GO:0000278; Fig. 2). The upregulated genes in the XI strains mostly involved cellular amino acid metabolic process, transmembrane transport, nucleobase-containing small molecule metabolic process (GO:0055086), vitamin metabolic process (GO:0006766), iron transport (GO:0006811), and ribosomal large subunit biogenesis (GO:0042273; Supplementary Materials 1: Table S2). The enhancement of the biosynthesis and transport of amino acids, nucleotides, and vitamins indicated that the expression of the XI pathway may cause some problem in amino acid, nucleobase, and vitamin metabolism. The downregulated genes mostly involved mitotic cell cycle, carbohydrate metabolic process (GO:0005975), chromatin organization (GO:0006325), regulation of organelle organization (GO:0033043), and transmembrane transport (Supplementary Materials 1: Table S3), which were closely related to the slower growth rate of the XI strains.

GO enrichment in different expressed genes between XI and XR–XDH strains. Only the top 20 GO terms with high gene coverage were listed. Up- or downregulated means the genes in XI strains up or downregulated compared with XR–XDH strain
Transcription Factors Analysis
The exhibition of complex genetic responses of xylose utilization strains was largely due to the transcription factors (TFs) that govern the flow of genetic information from DNA to mRNA [13, 14]. To explicate the regulation mechanism of different xylose utilization pathways, TF analysis was performed for the significantly different expressed genes. For the upregulated genes, Sfp1p, Ace2p, and Msn2p were the top three TF with the highest coverage of genes (Fig. 3). Sfp1p regulates transcription of ribosomal protein and biogenesis genes as well as response to nutrients and stress. It has been reported that SFP1 is involved in cell size modulation in response to nutrient conditions and its function is connected to protein kinase A (PKA) pathway [15]. Compared with XR–XDH strain, more than 85% of the upregulated genes in XI strains were regulated by Sfp1p (Fig. 3), which indicated that the insufficient carbon intake of XI strains caused cell starvation, and then induced Sfp1p to function. Meanwhile, the carbon metabolism could affect the cell growth and division, this might also be an important reason that Ace2p plays an important role in the regulation of upregulated (75.9%) and downregulated (84.7%) genes expression (Fig. 3), because Ace2p activated the genes required for cytokinesis and keep of cell wall integrity following cytokinesis [16]. Another TF Msn2p required by stress responses [17] covered 67.5% of the upregulated genes. This indicated that the XI strains showed a stronger stress response, probably because of lower xylose assimilating rates.

TF profiles regulated different genes expression in strains with different xylose metabolic pathways. Only the TFs with the coverage of genes higher than 60% were shown
For the downregulated genes, Ace2p, Tec1p, and Ste12p were the top three TF with the highest coverage of genes. Tec1p and Ste12p were required for haploid invasive and diploid pseudohyphal growth. The strains O7, P5, and Alpha25 were all haploid strains, which adopt elongated morphology in the absence of glucose (or other fermentable sugar) [18]. Because xylose is not a fermentable sugar for S. cerevisiae [19], the carbon starvation state faced by the xylose utilizing cells seemed complicated.
More importantly, both the Rap1p and Cst6p regulated 61.4% of the upregulated genes in XI strains. Rap1p plays a role in telomere structure; telomere is related to chromosome stability and cell cycle [20]. Cst6p is involved in the metabolism of non-optimal carbon sources [21] and chromosome stability. In this study, the xylA was multi-integrated in the δ region of the strain [9], but XYL1–XYL2 integrated in the URA3 region [7], whether the difference of integration sites would affect cell stability and then affect the xylose metabolism, is worth further study.
Central Carbon Metabolism
Xylose is metabolized to ethanol through the heterologous xylose utilization pathway, pentose phosphate pathway (PPP), glycolysis pathway, and alcohol fermentation pathway [22]. The expression of genes in the central carbon metabolism was investigated in detail in this study (Fig. 4).

Expression ratios of the genes involved in xylose metabolism pathway between XI and XR–XDH strains. The numbers indicated were log2(fold change). Red label means genes upregulated in both O7 and P7 compared with Alpha25; green label means genes downregulated in both O7 and P7 compared with Alpha25
Because lacking xylose-specific transporters, S. cerevisiae absorbed xylose mainly by non-specific hexose transporters of HXT family [4, 23]. From the results shown in Fig. 4, HXT2 and HXT4 were upregulated in the XI strains compared with those in the XR–XDH strain, whereas transcriptional levels of the putative hexose transporter genes HXT10 and HXT15 were downregulated. When xylose was the sole carbon source, HXT4 was one of the crucial sugar transporters [24, 25]. The differential expression of these genes indicated that the different xylose pathway might affect the xylose transport.
In the xylose metabolizing pathway, the conversion of xylulose to xylulose-5-phosphate catalyzed by xylulokinase is reported to be the rate-limiting step [22, 26]. Xylulokinase is encoded by XKS1, the expression level of which was downregulated in the XI strains. This may be related to the lower xylose consumption rates of the XI strains than the XR–XDH strain (Fig. 4).
In a non-oxidative PPP, transcriptional levels of key genes were downregulated in the XI strains compared with XR–XDH strain, including TKL1 and TAL1, which indicated a lower carbon flux in the XI strains, which was consistent with the previous study that the metabolites concentrations in the non-oxidative PPP of the XR–XDH strain were significantly higher than those in the XI strain [6]. Moreover, it is noteworthy that the expression level of RKI1 was upregulated in the XI strains. RKI1 encodes ribose-5-phosphate isomerase [23], which catalyzes the isomeration of ribulose-5-phosphate to ribose-5-phosphate (R5P) (Fig. 4). R5P serves as an important precursor metabolite for amino acid biosynthesis and nucleotide biosynthesis, and the expression level of the genes involved in the related amino acid and nucleotide biosynthesis pathway was also upregulated. In the oxidative PPP, the transcriptional level of GND1 was downregulated. GND1 encodes 6-phosphogluconate dehydrogenase [27], which catalyzes an NADPH regenerating reaction, and its downregulation might be because redox imbalance did not exist in the XI strains.
In glycolysis, the expression levels of the PFK1 (encoding 6-phosphofructokinase, catalyzed the production of fructose 1, 6-bisphosphate from fructose-6-phosphate) and PYK2 (encoding pyruvate kinase, catalyzed the production of pyruvate from phosphoenolpyruvate (PEP)) were downregulated in the XI strains (Fig. 4), which indicated that the carbon flux through glycolysis was assumed to be lower in the XI strains, because the elevated levels of PEP were recorded as a response to carbon starvation [28]. In the ethanol fermentation pathway, the expression level of PDC6, which encodes an isoform of pyruvate decarboxylase and has been identified to be important for xylose fermentation [29], was also downregulated in the XI strains.
Taken together, compared with the XR–XDH strain, the downregulated expression level of XKS1, PPP genes, glycolysis, and alcohol fermentation genes in the XI strains indicated that the carbon flux from xylulose to ethanol was lower in the XI strains, which directly reflected its lower xylose utilization rates.
Increased Expression of Purine and Amino Acid Biosynthesis Pathway in the XI Strains
Compared with the XR–XDH strain, the expression level of RKI1 was upregulated in the XI strains, indicating that the biosynthesis of R5P was affected by the xylA expression, whereas that of TKL1 and TAL1 was downregulated, indicating the reduced subsequent metabolism of R5P in non-oxidative PPP. Ribose-phosphate diphosphokinase catalyzes the transportation of pyrophosphate from ATP to R5P, generating 5-phospho-α-D-ribose 1-diphosphate (PRPP). PRPP is an important precursor in purine and pyrimidine nucleotide biosynthesis, cofactor biosynthesis, and amino acid biosynthesis. As shown in Fig. 5, PRPP can be metabolized to 5-amino-1-(5-phospho-β-D-ribosyl) imidazole (ARI) in five steps, and ARI is a key intermediate in the biosynthesis of purine nucleotides and thiamine. ARI can be further metabolized to inosine 5′-monophosphate (IMP). The initial three steps of this pathway were catalyzed by individual enzymes, whereas the last two steps were catalyzed by a single multifunctional enzyme, and the expression level of all the encoding genes was upregulated except ADE16. The upregulation of the majority of genes responsible for the biosynthesis from PRPP to adenosine monophosphate (AMP) may be propelled by a demand for the adenosine nucleotide pool [28].

Upregulation of purine nucleotide biosynthesis and amino acid metabolism. The numbers indicated were log2(fold change)
Consistent with the upregulated adenine nucleotide biosynthesis, the genes MTD1, SHM2, and GCV1 involved in the one-carbon metabolism using folate as the coenzyme were also upregulated. It is proposed that the one-carbon units from glycine were directed to purine biosynthesis and those from 5-methyl-THF (tetrahydrofolate) were directed to methionine biosynthesis [30]. Furthermore, the expression level of MET6, encoding homocysteine methyltransferase, was also upregulated. Serine is a co-substrate with THF for glycine and 5,10-methylene THF biosynthesis; the expression level of SER2 and SER33 in serine biosynthesis pathway was upregulated.
The expression of several other genes involved in amino acid metabolism was also upregulated, e.g., HIS4 and HIS7 responsible for histidine biosynthesis, ARG1 and ARG4 for arginine biosynthesis, and TRP4 and TRP5 for tryptophan biosynthesis. In addition to amino acid biosynthesis, many genes responsible for the transportation of amino acid were upregulated, e.g., for glutamate (AGP1, YMC2, ODC2, and DIP5), for lysine (AGP1 and ODC2), and aspartic acid (AGP1 and DIP5). These changes were regulated by Gcn4p and Sfp1p in response to amino acid starvation. The above results suggested that nucleobase and histidine were starved in XI strains, and the nucleobase and amino acid metabolism should be controlled in XI strains.
Decreased Expression of Biogenesis in the XI Strains
Compared with the XR–XDH strain, expression levels of the genes involved in the mitotic cell cycle were downregulated in the XI strains, which was consistent with the slower growth rates (Figs. 1 and 3). Moreover, genes expression involved in related cell processes such as chromatin organization and organelle organization were also downregulated (Supplementary Materials 1: Table S3). After cytokinesis, it is crucial to maintain cell wall integrity during mother–daughter cell separation to prevent cell lysis [31]; the corresponding genes involved in cell wall organization and biogenesis, such as CTS1 [16], PSA1 [32], YPS3/6 [33], and PIR3 [34], were downregulated, and this might also be caused by the carbon starvation. The cell wall is a vital cell organization essential for adaptation to stressful conditions [35, 36]; the downregulation of its biogenesis may indicate that the XI strains had a weaker tolerance to the inhibitors present in the lignocellulosic hydrolysate.
Conclusion
Although shared the same genetic background, the XR–XDH strain Alpha25 showed a higher xylose consumption rate than the XI strains O7 and P5. However, the strains O7 and P5 had higher ethanol yields than the strain Alpha25. Comparative transcriptome analysis indicated that compared with XR–XDH pathway, the XI pathway expression in industrial S. cerevisiae strain directly or indirectly affected the xylose metabolism. The GO analysis and TFs analysis revealed that after integrating xylA in the δ region; the DNA and chromosome stability and cell wall integrity of the S. cerevisiae strain were affected to some extent, which may be the reasons for the slow growth and fermentation of XI strain on xylose medium. The carbon starvation caused by lower xylose metabolism in XI strains further influenced the stress response and cell metabolism of amino acid, nucleobase, and vitamin. Besides, the genes involved in central carbon metabolism of XI strains were also affected by the xylA expression.
References
Zabed, H., Sahu, J. N., Boyce, A. N., & Faruq, G. (2016). Fuel ethanol production from lignocellulosic biomass: an overview on feedstocks and technological approaches. Renewable and Sustainable Energy Reviews, 66, 751–774.
Hoang, P. T. N., Ko, J. K., Gong, G., Um, Y., & Lee, S. M. (2018). Genomic and phenotypic characterization of a refactored xylose-utilizing Saccharomyces cerevisiae strain for lignocellulosic biofuel production. Biotechnology for Biofuels, 11(1), 268.
Cheng, C., Tang, R. Q., Xiong, L., Hector, R. E., Bai, F. W., & Zhao, X. Q. (2018). Association of improved oxidative stress tolerance and alleviation of glucose repression with superior xylose-utilization capability by a natural isolate of Saccharomyces cerevisiae. Biotechnology for Biofuels, 11(1), 28.
Cai, Z., Zhang, B., & Li, Y. (2012). Engineering Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: reflections and perspectives. Biotechnology Journal, 7(1), 34–46.
Demeke, M. M., Dietz, H., Li, Y., Foulquié-Moreno, M. R., Mutturi, S., Deprez, S., Den Abt, T., Bonini, B. M., Liden, G., Dumortier, F., Verplaetse, A., Boles, E., & Thevelein, J. M. (2013). Development of a D-xylose fermenting and inhibitor tolerant industrial Saccharomyces cerevisiae strain with high performance in lignocellulose hydrolysates using metabolic and evolutionary engineering. Biotechnology for Biofuels, 6(1), 89.
Bergdahl, B., Heer, D., Sauer, U., Hahn-Hägerdal, B., & van Niel, E. W. (2012). Dynamic metabolomics differentiates between carbon and energy starvation in recombinant Saccharomyces cerevisiae fermenting xylose. Biotechnology for Biofuels, 5(1), 34.
Li, Y.-C., Mitsumasu, K., Gou, Z.-X., Gou, M., Tang, Y.-Q., Li, G. Y., Wu, X. L., Akamatsu, T., Taguchi, H., & Kida, K. (2016). Xylose fermentation efficiency and inhibitor tolerance of the recombinant industrial Saccharomyces cerevisiae strain NAPX37. Applied Microbiology and Biotechnology, 100(3), 1531–1542.
Li, Y.-C., Li, G.-Y., Gou, M., Xia, Z. Y., Tang, Y.-Q., & Kida, K. (2016). Functional expression of xylose isomerase in flocculating industrial Saccharomyces cerevisiae strain for bioethanol production. Journal of Bioscience and Bioengineering, 121(6), 685–691.
Li, Y.-C., Zeng, W.-Y., Gou, M., Sun, Z. Y., Xia, Z.-Y., & Tang, Y.-Q. (2017). Transcriptome changes in adaptive evolution of xylose-fermenting industrial Saccharomyces cerevisiae strains with δ-integration of different xyla genes. Applied Microbiology and Biotechnology, 101, 1–13.
Wang, G., Tan, L., Sun, Z. Y., Gou, Z. X., Tang, Y. Q., & Kida, K. (2014). Production of bioethanol from rice straw by simultaneous saccharification and fermentation of whole pretreated slurry using Saccharomyces cerevisiae KF-7. Environmental Progress and Sustainable Energy, 34, 582–588.
Tang, Y., An, M., Liu, K., Nagai, S., Shigematsu, T., Morimura, S., & Kida, K. (2006). Ethanol production from acid hydrolysate of wood biomass using the flocculating yeast Saccharomyces cerevisiae strain KF-7. Process Biochemistry, 41(4), 909–914.
Gao, C., Fu, Q., Su, B., Zhou, S., Liu, F., Song, L., Zhang, M., Ren, Y., Dong, X., Tan, F., & Li, C. (2016). Transcriptomic profiling revealed the signatures of intestinal barrier alteration and pathogen entry in turbot (Scophthalmus maximus) following Vibrio anguillarum challenge. Developmental and Comparative Immunology, 65, 159–168.
Tan, S., & Richmond, T. J. (1998). Eukaryotic transcription factors. Current Opinion in Structural Biology, 8(1), 41–48.
Feng, X., & Zhao, H. (2013). Investigating host dependence of xylose utilization in recombinant Saccharomyces cerevisiae strains using RNA-Seq analysis. Biotechnology for Biofuels, 6(1), 96.
Cipollina, C., Alberghina, L., Porro, D., & Vai, M. (2005). SFP1 is involved in cell size modulation in respiro-fermentative growth conditions. Yeast, 22(5), 385–399.
King, L., & Butler, G. (1998). Ace2p, a regulator of CTS1 (chitinase) expression, affects pseudohyphal production in Saccharomyces cerevisiae. Current Genetics, 34(3), 183–191.
Schmitt, A. P., & Mcentee, K. (1996). Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences, 93(12), 5777–5782.
Cullen, P. J., & Sprague, G. F. (2000). Glucose depletion causes haploid invasive growth in yeast. PNAS, 97(25), 13619–13624.
Jin, Y. S., Laplaza, J. M., & Jeffries, T. W. (2004). Saccharomyces cerevisiae engineered for xylose metabolism exhibits a respiratory response. Applied and Environmental Microbiology, 70(11), 6816–6825.
Qun, Y., Runxiang, Q., Foland, T. B., Griesen, D., Gallowa, C. S., Chiu, Y. H., Sandmeier, J., Broach, J. R., & Bi, X. (2003). Rap1p and other transcriptional regulators can function in defining distinct domains of gene expression. Nucleic Acids Research, 31, 1224–1233.
Garcia-Gimeno, M. A., & Struhl, K. (2000). Aca1 and Aca2, ATF/CREB activators in Saccharomyces cerevisiae, are important for carbon source utilization but not the response to stress. Molecular and Cellular Biology, 20(12), 4340–4349.
Matsushika, A., Inoue, H., Kodaki, T., & Sawayama, S. (2009). Ethanol production from xylose in engineered Saccharomyces cerevisiae strains: current state and perspectives. Applied Microbiology and Biotechnology, 84(1), 37–53.
Matsushika, A., Goshima, T., & Hoshino, T. (2014). Transcription analysis of recombinant industrial and laboratory Saccharomyces cerevisiae strains reveals the molecular basis for fermentation of glucose and xylose. Microbial Cell Factories, 13(1), 16.
Sedlak, M., & Ho, N. W. Y. (2004). Characterization of the effectiveness of hexose transporters for transporting xylose during glucose and xylose co-fermentation by a recombinant Saccharomyces yeast. Yeast, 21(8), 671–684.
Zeng, W.-Y., Tang, Y.-Q., Gou, M., Xia, Z. Y., & Kida, K. (2016). Transcriptomes of a xylose-utilizing industrial flocculating Saccharomyces cerevisiae strain cultured in media containing different sugar sources. AMB Express, 6(1), 51.
Toivari, M. H., Aristidou, A., Ruohonen, L., & Penttilä, M. (2001). Conversion of xylose to ethanol by recombinant Saccharomyces cerevisiae: importance of xylulokinase (XKS1) and oxygen availability. Metabolic Engineering, 3(3), 236–249.
Ismail, K. S. K., Sakamoto, T., Hasunuma, T., & Kondo, A. (2013). Time-based comparative transcriptomics in engineered xylose-utilizing Saccharomyces cerevisiae identifies temperature-responsive genes during ethanol production. Journal of Industrial Microbiology and Biotechnology, 40(9), 1039–1050.
Kresnowati, M. T. A. P., van Winden, W. A., Almering, M. J. H., Ten Pierick, A., Ras, C., Knijnenburg, T. A., Daran-Lapujade, P., Pronk, J. T., Heijnen, J. J., & Daran, J. M. (2006). When transcriptome meets metabolome: Fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology, 2, 16.
Zeng, W.-Y., Tang, Y.-Q., Gou, M., Sun, Z. Y., Xia, Z. Y., & Kida, K. (2017). Comparative transcriptomes reveal novel evolutionary strategies adopted by Saccharomyces cerevisiae with improved xylose utilization capability. Applied Microbiology and Biotechnology, 101(4), 1753–1767.
Piper, M. D., Hong, S. P., Ball, G. E., & Dawes, I. W. (2000). Regulation of the balance of one-carbon metabolism in Saccharomyces cerevisiae. The Journal of Biological Chemistry, 275(40), 30987–30995.
Frýdlová, I., Malcová, I., Vašicová, P., & Hašek, J. (2009). Deregulation of DSE1 gene expression results in aberrant budding within the birth scar and cell wall integrity pathway activation in Saccharomyces cerevisiae. Eukaryotic Cell, 8(4), 586–594.
Janik, A., Sosnowska, M., Kruszewska, J., Krotkiewski, H., Lehle, L., & Palamarczyk, G. (2003). Overexpression of GDP-mannose pyrophosphorylase in Saccharomyces cerevisiae corrects defects in dolichol-linked saccharide formation and protein glycosylation. Biochimica et Biophysica Acta, 1621(1), 22–30.
Krysan, D. J., Ting, E. L., Abeijon, C., Kroos, L., & Fuller, R. S. (2005). Yapsins are a family of aspartyl proteases required for cell wall integrity in Saccharomyces cerevisiae. Eukaryotic Cell, 4(8), 1364–1374.
Sumita, T., Yoko-o, T., Shimma, Y., & Jigami, Y. (2005). Comparison of cell wall localization among pir family proteins and functional dissection of the region required for cell wall binding and bud scar recruitment of Pir1p. Eukaryotic Cell, 4(11), 1872–1881.
Klis, F. M., Boorsma, A., & De Groot, P. W. (2006). Cell wall construction in Saccharomyces cerevisiae. Yeast, 23(3), 185–202.
de Lucena, R. M., Elsztein, C., de Barros Pita, W., de Souza, R. B., de Sá Leitão Paiva Júnior, S., & de Morais Junior, M. A. (2015). Transcriptomic response of Saccharomyces cerevisiae for its adaptation to sulphuric acid-induced stress. Antonie Van Leeuwenhoek, 108(5), 1147–1160.
Funding
This work was supported by the National Natural Science Foundation of China (31170093), the Talent Project for Science and Technology Innovation of Sichuan Province (2017RZ0021), and the Open Fund of Key Laboratory of Development and Application of Rural Renewable Energy, Ministry of Agriculture of China (2018-009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, YC., Xie, CY., Yang, BX. et al. Comparative Transcriptome Analysis of Recombinant Industrial Saccharomyces cerevisiae Strains with Different Xylose Utilization Pathways. Appl Biochem Biotechnol 189, 1007–1019 (2019). https://doi.org/10.1007/s12010-019-03060-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-019-03060-8