
Abstract
Valyl-glycine (Val-Gly) is useful as a synthetic substrate of γ-glutamyl-valyl-glycine (γ-Glu-Val-Gly), which exhibits a strong taste of “kokumi.” For efficient enzymatic synthesis of Val-Gly from valine methylester and glycine using L-amino acid esterase (LAE), we screened microorganisms producing LAE with synthetic activity toward Val-Gly. Among 17 isolates showing LAE activity, Elizabethkingia sp. TT1, which was identified by 16S rDNA sequence analysis, showed the highest synthetic activity toward Val-Gly. LAE from Elizabethkingia sp. TT1 (TT1LAE) was purified approximately 1300 times, resulting in a yield of 2.8% and specific activity of 118.8 μmol/min/mg protein. SDS-PAGE analysis revealed a subunit molecular mass of 78 kDa. The molecular mass of the native enzyme determined by gel filtration was 103 kDa. The purified enzyme showed maximum activity at pH 9.0 and at a temperature of 25 °C, and it was stable over the pH range of 5.0–8.5 and 25 °C–40 °C. No metal ions that were tested had a significant effect on enzyme activity, but the enzyme was slightly inhibited by EDTA.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dipeptides are composed of two amino acids linked by a peptide bond, and they possess useful properties. For example, the solubility or the stability of L-glutamine in aqueous solution can be improved by its conversion to alanyl-glutamine (Ala-Gln) [1], valyl-tyrosine (Val-Tyr) decreases blood pressure [2], seryl-histidine (Ser-His) has an analgesic effect [3], L-arginyl dipeptides are salt taste enhancer [4], and β-alanyl-histidine (β-Ala-His) shows antioxidant activity [5].
Among various useful dipeptides, valyl-glycine (Val-Gly) is known to be useful as a synthetic substrate of γ-glutamyl-valyl-glycine (γ-Glu-Val-Gly). γ-Glu-Val-Gly has recently received increased attention as a novel food additive, because it has a strong taste of kokumi [6]. Kokumi enhances the five basic tastes, particularly sweet, salty, and umami, and modifies the thickness and richness of food [7]. Among several kokumi substances, γ-Glu-Val-Gly has been reported to be a strong kokumi peptide, and the sensory activity of this kokumi substance was found to be 12.8-fold greater than that of glutathione [8]. γ-Glu-Val-Gly has been reported to be present in several foods such as scallops, fermented fish sauces, and soy sauces [9–11]. In contrast, γ-Glu-Val-Gly can be effectively produced by γ-glutamyltransferase (GGT) from Val-Gly and γ-glutamyl compounds. The γ-Glu-Val-Gly synthetic activity of GGT derived from Escherichia coli and Pseudomonas sp. has been reported [6].
In general, several methods have been reported for the dipeptide synthesis. Chemical syntheses involve liquid-phase peptide synthesis via N-carboxyanhydride intermediate and a synthetic process using D-2-chloropropoinyl-amino acid [12, 13]. These methods require many complicated steps, such as substrate protection and de-protection of the product protection group. Alternatively, enzymatic synthesis can be employed for dipeptide synthesis. Ala-Gln production by direct fermentation using recombinant E. coli expressing L-amino acid ligase (Lal) and L-amino acid esterase (LAE, α-amino acid ester acyltransferase (AET)) has been reported [14, 15]. Because the former is a fermentation method using glucose and ammonia as the main raw materials, it is a cost-effective method for dipeptide production, but it has disadvantages, including the low accumulation and efficiency of the required dipeptide. The latter is an excellent method of dipeptide production, because the Ala-Gln production yield is 67% against substrates [15]. However, there have been few reports on the enzymatic synthesis of a dipeptide using LAE and no reports on the enzymatic synthesis of Val-Gly. In this study, we focused on the enzymatic synthesis of Val-Gly.
To establish the method for enzymatic synthesis of Val-Gly from valine methylester and glycine using LAE (Fig. 1), microorganisms exhibiting LAE activity were screened from the soil. As a result, among 17 microorganisms isolated, Elizabethkingia sp. TT1 exhibited the highest synthetic activity of Val-Gly using valine methylester and glycine as substrates, followed by Pseudomonas putida and Stenotrophomonas maltophilia. Here, we have described the production, purification, and characterization of LAE from Elizabethkingia sp. TT1 (TT1LAE).

Synthetic reaction of Val-Gly by LAE
Materials and Methods
Chemicals
Reagent-grade chemicals were purchased from Wako Pure Chemical Industries, Japan, unless stated otherwise.
Media
Medium 1 contained 0.2% Val-OMe, 0.05% KH2PO4, 0.15% K2HPO4, and 0.05% MgSO4·7H2O (pH 7.0). Medium 2 contained 0.5% D-Glucose, 0.5% yeast extract, 0.5% polypepton, 0.1% MgSO4·7H2O, and 1.5% agar (pH 7.0). Medium 3 contained 0.5% D-glucose, 0.5% yeast extract, 0.5% polypepton, 0.2% (NH4)2SO4, 0.05% KH2PO4, 0.15% K2HPO4, and 0.05% MgSO4·7H2O (pH 7.0).
Screening Methods
After liquid medium 1 was incubated with soil samples for 2 or 3 days at 30 °C, each culture was subcultured in liquid medium 1. The subcultured samples were streaked on solid medium 2. Colonies of the isolates were inoculated in a test tube containing 5 ml of medium 3, followed by cultivation under 250 rpm at 30 °C for 18–20 h. Thereafter, the cells were harvested by centrifugation (4 °C, 10,000×g, 5 min) to obtain intact cells. The reaction was conducted at 30 °C for 2 h using a reaction solution containing 100 mM L-valine methylester (Val-OMe), 500 mM glycine (Gly) (Nacalai tesque, Kyoto, Japan) and 10 mM EDTA (Nacalai tesque, Kyoto, Japan) as well as intact cells in 100 mM borate buffer (pH 9.0). The Val-Gly produced was qualitatively and quantitatively measured as described in the “Analysis” section.
16S rDNA Sequence Analysis
The 16S rDNA sequence of the isolates with relatively high Val-Gly synthetic activity was determined by directly sequencing a PCR product of the 16S rDNA gene. The gene was amplified using Blend Taq DNA polymerase (TOYOBO, Osaka, Japan), primers 20f (5′-TGTAAATCGGCCAGTAGAGTTTGATCCTGGCTC-3′) and 1510r (5′-CAGGAAACAGCTATGACCGGCTACCTTGTTACGACT-3′), and chromosomal DNA. DNA sequence similarity searches were performed using the GenBank BLAST program (http://www.ncbi.nlm.nih.gov).
Cultivation of Elizabethkingia Sp. TT1
To determine the optimal culture conditions for LAE production, Elizabethkingia sp. TT1 was cultivated in a medium containing the carbon and nitrogen sources (0.05–0.5%), 0.15% K2HPO4, 0.1% KH2PO4, and 0.05% MgSO4·7H2O by shaking at 30 °C for 24 h.
Enzyme Purification
All purification steps were performed at 4–8 °C unless otherwise stated. The buffer used was 10 mM potassium phosphate buffer (pH 7.0, buffer I). The harvested cells were washed twice with buffer I and collected by centrifugation (16,890×g) at 4 °C for 15 min. The washed cells were suspended in buffer I and disrupted by sonication at 4–8 °C for 15 min. The cell debris was removed by centrifugation (16,890×g) at 4 °C for 15 min. The supernatant was subjected to ammonium sulfate precipitation between 40 and 70% saturation. The precipitate was then recovered by centrifugation (16,890×g) at 4 °C for 15 min, resuspended in a minimal volume of buffer I, and dialyzed against buffer I at least twice for 16 h. The dialyzed enzyme solution was applied to a CM-Cellulofine (Seikagakukogyo, Tokyo, Japan) column equilibrated with buffer I. After washing the column with buffer I, the enzyme was eluted with buffer I containing 0.05 M NaCl. The active fractions were collected and dialyzed against buffer I. Solid ammonium sulfate was added to the enzyme solution to 30% saturation and applied to a Butyl-Cellufine (JNC, Tokyo, Japan) column equilibrated with the buffer containing ammonium sulfate to 30% saturation. After washing the column with the same buffer, the enzyme was eluted with a linear salt down gradient of 30–20% saturated ammonium sulfate in buffer I. The active fractions were collected and dialyzed against buffer I. Solid ammonium sulfate was added to the enzyme solution to 30% saturation and applied to the Butyl-FF (GE Healthcare Japan, Tokyo, Japan) column equilibrated with the buffer containing ammonium sulfate at 30% saturation. After washing the column with the same buffer, the enzyme was eluted with a linear salt down gradient of 30–20% saturated ammonium sulfate in buffer I. The active fractions were collected and dialyzed against buffer I. The enzyme was applied to a Superdex 200 HR 10/30 (GE Healthcare Japan, Tokyo, Japan) gel filtration column and ran with 50 mM potassium phosphate buffer supplemented with 150 mM NaCl. The active fractions were collected and dialyzed against buffer I. The enzyme preparation was concentrated by ultrafiltration and stored at 4 °C.
Substrate Specificity of TT1LAE for Hydrolysis Reaction
The LAE activity was assayed by determining the concentration of released methanol. A reaction mixture containing 0.25 U alcohol oxidase (Sigma-Aldrich Japan, Tokyo, Japan), 1 U horseradish peroxidase, and 1 mM o-phenylenediamine in 0.1 M potassium phosphate buffer (pH 7.0) was prepared for the detection of methanol. To the reaction mixture, 10 μl of LAE reaction solution was added, and the reaction mixture was incubated at 30 °C for 30 min. The absorbance at 420 nm was measured. To investigate specificity for amino acid methyl ester, the reaction was conducted at 30 °C for 15 min in the solution containing 60 mM L-amino acid methyl ester (pH 9.0).
Analysis
The protein concentration was determined by the Lowry method using egg albumin as the standard [16]. The molecular mass of the denatured enzyme was estimated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) [17], and the gel was stained with Coomassie Brilliant Blue (Nacalai tesque, Kyoto, Japan). Val-Gly was qualitatively measured by paper chromatography using chromatography paper (ADVANTEC, Tokyo, Japan) (46 cm × 14 cm) using n-butanol/acetic acid/water (4:1:1, v/v) as the solvent. Val-Gly was visualized by heating the chromatography paper treated with 0.1% 2,4,6-trinitrobenzenesulfonic acid sodium salt dihydrate (TNBS) and 0.125 M Na2B4O7 (Nacalai tesque, Kyoto, Japan) solution. Val-Gly was quantitatively measured by high-performance liquid chromatography using a COSMOSIL 5C18-MS-II column (4.6 × 250 mm) (Nacalai tesque, Kyoto, Japan) equilibrated with 0.1 M acetate buffer (pH 6.0) containing 7% acetonitrile and 3% tetrahydrofuran. The derivatized Val-Gly was eluted using a linear gradient of acetonitrile (Nacalai tesque, Kyoto, Japan) (7–47%) at a flow rate of 1 ml/min. The standard reaction mixture for Val-Gly synthesis contained 100 mM Tris-HCl buffer (pH 9.0), 70 mM L-Val-OMe, 500 mM Gly (pH 9.0), and the enzyme at a final volume of 0.1 ml. After a reaction time of 15–30 min at 25 °C, the reaction was terminated by boiling for 10 min. One unit of enzyme activity was defined as the amount of the enzyme that produced 1 μmol of Val-Gly per minute. For matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) analysis, the reaction mixture for Val-Gly synthesis was diluted 50-fold with Milli Q water. One microliter of the sample was mixed with 1 μl of matrix solution on a plate. The plate was dried and loaded into MALDI-TOF MS (autoflex speed, Bruker Daltonics K.K., Kanagawa, Japan). The matrix (2,5-dihydroxybenzoic acid, DHB) was dissolved in the solvent (0.1% trifluoroacetic acid/acetonitrile = 1:2).
Results
Screening of Microorganisms Showing Val-Gly Synthetic Activity
In total, 315 isolates were tested for their Val-Gly production abilities from L-Val-OMe and Gly in an aqueous solution. After screening, 17 strains were selected as Val-Gly producers. Among these, 7 strains exhibiting higher Val-Gly synthetic activity were identified by analyzing their 16S ribosomal RNA (rRNA) gene sequences. As shown in Fig. 2, Elizabethkingia sp., Stenotrophomonas maltrophilia, and several species of Pseudomonas were dominantly isolated. Among these isolates, Elizabethkingia sp. TT1 showed the highest activity. Empedobacter brevis and Sphingobacterium siyangensis have been reported to show high Ala-Gln synthetic activity [17]. The strain TT1 also showed high Ala-Gln synthetic activity (data not shown). Phylogenetic analysis indicated that the strain TT1 was closer relatives of Empedobacter and Sphingobacterium than those of Stenotrophomonas and Pseudomonas.

16S rRNA sequence phylogenetic tree. The tree was constructed using the neighbor-joining method. The percentage of replicate trees in which the associated strains were clustered together in the bootstrap test was shown next to the branches. TT1, 2, 3, 4, 6, 7, and 8 strains were found in this study. Dipeptide synthesis by LAE was reported in E. brevis and S. siyangensis [21] while Ala-Gln synsthesis was reported in P. putida [20]
Effects of Carbon and Nitrogen Sources on LAE Production
The effects of various carbon and nitrogen sources on LAE production were examined (Table 1). A medium containing polypeptone provided relatively high cell growth and total LAE activity, but resulted in low specific activity. The highest specific activity (0.082 U/mg) was obtained when glucose and casamino acids were used as carbon source and nitrogen sources, respectively.
Enzyme Purification
The enzyme was purified from Elizabethkingia sp. TT1 using the following purification steps: ammonium sulfate fractionation, cation exchange using CM-Cellulofine, hydrophobic interaction using Butyl-Cellufine and Butyl-FF, and gel filtration using Superdex 200 HR 10/30. The overall enzyme purification was 1300.1-fold with a yield of 2.8% (Table 2). The purified enzyme was considered to be homogeneous by SDS-PAGE (Fig. 3). The apparent molecular mass of the purified enzyme was calculated to be 103 kDa by gel filtration (Superdex 200 HR 10/30). The molecular mass of the denatured enzyme was determined to be 78 kDa by SDS-PAGE.

SDS-PAGE of purified TT1LAE. Lanes: 1 molecular markers, 2 cell-free extract, 3 ammonium sulfate fractionation, 4 CM-cellulofine, 5 Butyl-Cellufine, 6 Butyl-FF, 7 purified TT1LAE (Superdex 200 HR 10/30), 8 molecular markers
Enzyme Characterization
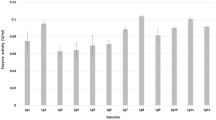
The enzyme showed the highest activity at pH 9.0. It was most stable at pH 8.0 and relatively stable in an acidic environment (Fig. 4). The optimum temperature was approximately at 25 °C, and the enzyme retained 80% of its activity following treatment at 40 °C for 10 min (Fig. 5). Most of the metal ions tested had little effect on enzyme activity, but the enzyme was slightly inhibited by EDTA (Table 3). In addition, Val-Gly synthetic activity of TT1LAE was strongly inhibited by phenylmethanesulfonyl fluoride (5 mM, PMSF). Substrate specificity for hydrolysis reaction was examined by a colorimetric method. The results showed that nonbulky amino acid methyl esters, such as Ala-OMe and Gly-OMe, were preferred over Val-OMe as acyl donors (Fig. 6).

Effect of pH on a activity and b stability of purified TT1LAE. Enzyme activity was assayed at various pH values using the following buffers: citrate buffer (pH 4–6; empty circles), potassium phosphate buffer (pH 6–8; filled squares), Tris-HCl buffer (pH 8–9; filled triangles), and borate buffer (pH 9–10.5; filled circles). For pH stability testing, the purified enzyme was incubated at various pH values at 4 °C for 12 h and residual activity was measured under standard conditions. All the tests were performed in duplicates

Effect of temperature on a activity and b stability of purified TT1LAE. Enzyme activity was assayed at various temperatures under standard reaction conditions. For thermal stability testing, the purified enzyme was incubated at various temperatures for 10 min, and residual activity was measured under standard conditions. All the tests were performed in duplicates

Substrate specificity of TT1LAE for hydrolysis reaction. The LAE activity was assayed by determining the concentration of released methanol. Each error bar indicates the standard deviation for a triplicate analysis
Analysis of the Reaction Products
The reaction mixture was analyzed by using MALDI-TOF MS. The peaks at m/z 175, 197, and 213, which correspond to [Val-Gly + H]+, [Val-Gly + Na]+, and [Val-Gly + K]+, were detected, indicating the formation of Val-Gly under the reaction condition (Fig. 7). The synthesis of Val-Gly was also confirmed by HPLC analysis (20.7 mM). On the other hand, L-Val was detected by paper chromatography as a by-product (data not shown). However, no mass peaks corresponding to the formation of Val-Val-OMe were observed. This result suggested that Val-Val-OMe could be scarcely produced or that its concentration in the reaction mixture might be below detection limit of MALDI-TOF MS.

MALDI-TOFMS analysis of the reaction catalyzed by TT1LAE. Peaks assigned are as follows: m/z 175, [Val-Gly + H]+ ; m/z 197, [Val-Gly + Na]+ ; m/z 213, [Val-Gly + K]+
Discussion
Several enzymatic methods for dipeptide synthesis have been reported. The first documented method is peptidase-catalyzed synthesis. Some methods for peptidase-catalyzed synthesis have been reported. Hatanaka et al. reported the syntheses of the dipeptides, aspartyl-phenylalanine, alanyl-tyrosine, and valyl-tyrosine methyl esters using free amino acids as acyl donors and aminoacyl methyl esters as acyl acceptors in 98% methanol (MeOH) in addition to aminopeptidase from Streptomyces septatus TH-2 [18]. Ariyoshi et al. reported the syntheses of the dipeptide esters, (R)-Ama-(S)-Phe-OMe and (R)-Ama-(S)-Phe-OEt by the coupling of N-protected L-amino acids and C-protected L-amino acids using thermolysin [19]. These methods still lack industrial requirements in terms of many complicated steps such as protection of the substrate and de-protection of the product protection group and low yield and productivity. There have been no reports on Val-Gly synthesis using peptidases.
The second method of dipeptide synthesis involves Lal-catalyzed reactions. Lal synthesizes dipeptides from unprotected L-amino acids in an ATP-dependent manner [20]. Lal is useful for efficient dipeptide synthesis because the substrates are unprotected amino acids. Therefore, several Lals have been investigated, and synthesis of many kinds of dipeptides such as Ala-Gln, Phe-Ala, Met-Gly, Leu-Gly, and D-Ala-Ala has been reported [20–23]. However, it has disadvantages such as low accumulation and productivity of the required dipeptides. There have been no reports on Val-Gly synthesis using Lal.
LAE-catalyzed synthesis of dipeptides is a very simple and highly productive method [15]. Furthermore, kilo-scale quantities of dipeptides have been reported to be produced using recombinant E. coli expressing LAE [15]. Therefore, it is very efficient for the industrial production of dipeptides. Because this is the first report of enzymatic synthesis of Val-Gly using LAE, it has important implications for the enzymology and industrial applications of Val-Gly.
TT1LAE may be closely homologous to the reported LAE from E. brevis ATCC14234 (EBLAE) and S. siyangensis AJ2458 (SSLAE) [24, 25]. The molecular size (TT1LAE, 105 kDa; EBLAE, 150 kDa; and SSLAE, 115 kDa), optimal temperature (TT1LAE, 25 °C; EBLAE, 30 °C; and SSLAE, 20 °C), and optimal pH (TT1LAE, pH 9.0; EBLAE, pH 8.5; and SSLAE, pH 8.5) of these were similar.
Hydrolytic activity assay of the LAE showed that TT1LAE had strong substrate preference to nonbulky amino acid methyl esters as the donor substrate, and Ala-OMe appeared to be most preferred. Unfortunately, due to the lack of the standard dipeptides in our laboratory, the preference of TT1LAE to the acceptor substrate could not be evaluated in this study. Examination of the acceptor preference of the enzyme will be a future task since it is useful information for the synthesis of other valyl derivatives.
It is expected that TT1LAE is a constitutively active enzyme as the enzyme showed the highest specific activity when glucose and casamino acids were used as carbon source and nitrogen sources, respectively, and the specific activity did not increase when Val-OMe was used. Because TT1LAE was hardly affected by most metals and EDTA and was inhibited by PMSF, it is suggested that TT1LAE is a nonmetalloenzyme and a kind of serine-peptidase. One of the applications of Val-Gly is the production of γ-Glu-Val-Gly [6]. We have validated that GGT derived from Pseudomonas sp. can synthesize γ-Glu-Val-Gly from Val-Gly and glutathione with a high yield (unpublished data). It is, therefore, expected that γ-Glu-Val-Gly can be effectively produced by a fermentation method using recombinant E. coli expressing TT1LAE and GGT from Pseudomonas sp. However, further investigations are required to improve the industrial applications of the enzyme, for example, cloning of the gene that produces the enzyme and improvement of the thermal stability. Further experiments are in progress to clone the gene of TT1LAE. Whole amino acid sequence of the enzyme, which is important information from the applied enzymology point of view, will be clarified on the basis of nucleotide sequence information.
In conclusion, we found a novel LAE from Elizabethkingia sp. TT1. Because TT1LAE generates Val-Gly with yields exceeding 40% relative to Val-OMe without energy sources and metal ions, the enzyme is useful for Val-Gly production.
References
Kweon, M. N., Moriguchi, S., Mukai, K., & Kishino, Y. (1991). Effect of alanylglutamine-enriched infusion on tumor growth and cellular immune function in rats. Amino Acids, 1, 7–16.
Lieselot, V., Nicole, M., John, V. C., Justyna, S., & Guy, S. (2008). Antihypertensive mechanism of the dipeptide Val-Tyr in rat aorta. Peptides, 29, 261–267.
Asechi, M., Tomonaga, S., Tachibana, T., Han, L., Hayamizu, K., Michael, D. D., et al. (2006). Intracerebroventricular injection of l-serine analogs and derivatives induces sedative and hypnotic effects under an acute stressful condition in neonatal chicks. Behavioural Brain Research, 170, 71–77.
Harth, L., Krah, U., Linke, D., Dunkel, A., Hofmann, T., & Berger, R. G. (2016). Salt taste enhancing l-arginyl dipeptides from casein and lysozyme released by peptidases of basidiomycota. Agricultural and Food Chemistry.
Kohen, R., Yamamoto, Y., Cundy, K. C., & Ames, B. N. (1988). Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proceedings of the National Academy of Sciences of the United States of America, 85, 3175–3179.
Nozaki, H., Abe, I., Takakura, J., Takeshita, R., Suzuki, H., & Suzuki, H., Japan Patent Kokai 2013–075908 (2013.04.11).
Kuroda, M., Yamanaka, T., & Miyamura, N. (2004). Change in taste and flavor of food during the aging with heating process. Generation of “KOKUMI” flavor during the heating of beef soup and beef extract. Japanese journal of Taste and Smell Research, 11, 175–180.
Ohsu, T., Amino, Y., Nagasaki, H., Yamanaka, T., Takeshita, S., Hatanaka, T., et al. (2010). Involvement of the calcium-sensing receptor in human taste perception. Biological Chemistry, 285, 1016–1022.
Kutoda, M., Kato, Y., Yamazaki, J., Kageyama, N., Mizukoshi, T., Miyano, H., et al. (2012). Determination of γ-glutamyl-valyl-glycine in raw scallop and processed scallop products using high pressure liquid chromatography-tandem mass spectrometry. Food Chemistry, 134, 1640–1644.
Kuroda, M., Kato, Y., Yamazaki, J., Kai, Y., Mizukoshi, T., Miyano, H., et al. (2012). Determination and quantification of γ-glutamyl-valyl-glycine in commercial fish sauces. Japanese journal of Food Chemistry, 60, 7291–7296.
Kuroda, M., Kato, Y., Yamazaki, J., Kai, Y., Mizukoshi, T., Miyano, H., et al. (2013). Determination and quantification of the kokumi peptide, γ-glutamyl-valyl-glycine, in commercial soy sauces. Food Chemistry, 141, 823–828.
John, C. S., & Yang, D. H. (1958). The use of N-formylamino acids in peptide synthesis. American Chemical Society, 80, 1154–1158.
Eric, M. G., Langlois, J. R., & Williams, R. E. (1973). Use of N-Carboxyanhydrides in the synthesis of dipeptides. Factors affecting the yields of several alanine and valine-containing dipeptides. Canadian Journal of Chemistry, 51, 1284–1287.
Tabata, K., & Hashimoto, S. (2007). Fermentative production of L-alanyl-L-glutamine by a metabolically engineered Escherichia coli strain expressing L-amino acid α-ligase. Applied and Environmental Microbiology, 73, 6378–6385.
Hirao, Y., Mihara, Y., Kira, I., Abe, I., & Yokozeki, K. (2013). Enzymatic production of L-alanyl-L-glutamine by recombinant E. coli from Sphingobacterium siyangensis. Bioscience, Biotechnology, and Biochemistry, 77, 618–623.
Lowry, O., Rosebrough, N., Farr, A., & Randall, R. (1951). Protein measurement with the folin phenol reagent. Biological Chemistry, 193, 265–275.
Laemmli, U. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Arima, J., Uesugi, Y., Uraji, M., Iwabuchi, M., & Hatanaka, T. (2006). Dipeptide synthesis by an aminopeptidase from Streptomyces septatus TH-2 and its application to synthesis of biologically active peptides. Applied and Environmental Microbiology, 72, 4225–4231.
Ota, M., Nio, N., & Ariyoshi, Y. (1993). Enzymatic synthesis and chemical properties of sweet aminomalonyl (Ama) dipeptide ester (R)-Ama-(S)-Phe-OMe and (R)-Ama-(S)-Phe-OEt. Bioscience, Biotechnology, and Biochemistry, 57, 808–813.
Tabata, K., Ikeda, H., & Hashimoto, S. (2005). ywfE in Bacillus subtilis codes for a novel enzyme, L-amino acid ligase. Bacteriology, 187, 5195–5202.
Kino, K., Nakazawa, Y., & Yagasaki, M. (2008). Dipeptide synthesis by L-amino acid ligase from Ralstonia solanacearum. Biochemical and Biophysical Research Communications, 371, 536–540.
Tran, H. T., Hong, M. K., Ngo, H. P. T., Huynh, K. H., Ahn, Y. J., Wang, Z., & Kang, L. W. (2016). Structure of d-alanine-d-alanine ligase from Yersinia pestis: nucleotide phosphate recognition by the serine loop. Acta Crystallographica Section D: Structural Biology, 72(1), 12–21.
Kino, K., Noguchi, A., Nakazawa, Y., & Yagasaki, M. (2008). A novel L-amino acid ligase from Bacillus licheniformis. Bioscience and Bioengineering, 106, 313–315.
Yokozeki, K., & Hara, S. (2005). A novel and efficient enzymatic method for the production of peptides from unprotected starting materials. Biotechnology, 115, 211–220.
Abe, I., Hara, S., & Yokozeki, K. (2011). Gene cloning and characterization of α-amino acid ester acyl transferase in Empedobacter brevis ATCC14234 and Sphingobacterium siyangensis AJ2458. Bioscience, Biotechnology, and Biochemistry, 75, 2087–2092.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tanaka, T., Takagi, K., Saddam, H.M. et al. Purification and Characterization of Elizabethkingia L-Amino Acid Esterase: an Enzyme Useful for Enzymatic Synthesis of the Dipeptide, Valyl-Glycine. Appl Biochem Biotechnol 183, 362–373 (2017). https://doi.org/10.1007/s12010-017-2450-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2450-3