
Abstract
WRKY transcription factors belong to a large family of plant transcriptional regulators whose members have been reported to be involved in a wide range of biological roles including plant development, adaptation to environmental constraints and response to several diseases. However, little or poor information is available about WRKY’s in Citrus. The recent release of completely assembled genomes sequences of Citrus sinensis and Citrus clementina and the availability of ESTs sequences from other citrus species allowed us to perform a genome survey for Citrus WRKY proteins. In the present study, we identified 100 WRKY members from C. sinensis (51), C. clementina (48) and Citrus unshiu (1), and analyzed their chromosomal distribution, gene structure, gene duplication, syntenic relation and phylogenetic analysis. A phylogenetic tree of 100 Citrus WRKY sequences with their orthologs from Arabidopsis has distinguished seven groups. The CsWRKY genes were distributed across all ten sweet orange chromosomes. A comprehensive approach and an integrative analysis of Citrus WRKY gene expression revealed variable profiles of expression within tissues and stress conditions indicating functional diversification. Thus, candidate Citrus WRKY genes have been proposed as potentially involved in fruit acidification, essential oil biosynthesis and abiotic/biotic stress tolerance. Our results provided essential prerequisites for further WRKY genes cloning and functional analysis with an aim of citrus crop improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Citrus is one of the world’s most and widest cultivated crop [1] with a worldwide production of 135 million tons in 2013 [2]. Because of their widespread geographical distribution and being thus cultivated under variable pedo-climatic conditions, citrus crops face abiotic and biotic stresses that damage final production and depreciate fruit quality. At the genomic level, Citrus and related genera have 18 chromosomes in diploid somatic cells [3]. Besides, there is a financial market pressure and a constant demand from the industries for better quality products in order to meet the consumers’ needs and expectations. Thereby, launching Citrus breeding and improvement program has become a major goal and a priority for both farmer producers and industrial companies [4]. Thus, based on the fact that the only way to propagate citrus cultivars is by grafting onto rootstock, scientists from concerned countries focused their research efforts on selecting and combining rootstocks traits of abiotic (salinity and drought) and biotic (mostly Citrus Tristeza Virus CTV and Phytophthora) stresses tolerance. Such interesting and useful traits of tolerance and acclimatization could be found within Citrus and their relatives [5]. However, due to major constraints related to the Citrus biology of reproduction such as high level of heterozygosity, sterility of several varieties, apomixis or nucellar polyembryony phenomena and mostly self-incompatibility within Citrus cultivars, breeding of the species by conventional procedures through sexual crossing has limited success. The recent advent of new biotechnological tools has opened new insights and promising prospects for Citrus improvement, making those limitations bypassed [6].
Citrus breeding community has thus focused efforts on newly released genomic resources and transcriptomic data from international sequencing centers and programs. Interestingly, recent release of two Citrus species genome sequences provides an opportunity to explore molecular data and resources for Citrus breeding program [7]. ‘Clementine’ mandarin genome was the first citrus sequenced genome, serving as genome of reference for citrus [8] and was followed by ‘Ridge Pineapple’ sweet orange clone using next generation sequencing. These projects load a draft of high quality assemblage [4].
In the past 10 years, several work projects related to Citrus omics data and resources have emerged [9–13]. Yet, to reach the breeding aims and objectives, efforts still to do by combining genomic technology and genes of agricultural interest identification and manipulation [13].
Gene expression regulation is a vital mechanism for all living organisms. Transcription factors (TF) and other related components together act in the transcriptional regulating network, through binding the specific DNA sequence and activating and/or repressing the transcription of their target genes. Among these, WRKY proteins are considered important transcriptional regulators in defense signaling. The WRKY TFs family is one of the largest families of plant TFs which has been characterized for several and diverse plants species [14–16]. Usually, the WRKY protein has one or two WRKY domains of nearly 60 aa containing at its N-terminus the WRKYGQK conserved motif and a zinc finger motif at its C-terminus [15, 17, 18]. The WRKYGQK aa peptide displays a β-strand structure that directly binds to DNA on the regulatory region of target genes. The WRKY binding site is made of a conserved W box: (C/T) TGAC (T/C) which is frequently found within promoter region of stress response and development associated genes. Group I WRKY TF contains two WRKY domains and is thought to be ancestral type [15] while genes containing a single WRKY domain are classified into groups II (a, b, c, d or e) or III according to their aa sequence and the structure of their zinc finger motifs [15, 19]. Rushton et al. [17] were first to report that WRKYs are involved in plant defense. In fact, WRKYs are also implicated in plant growth regulation and development such as seed dormancy and development, germination, rhizogenesis, morphogenesis of trichomes, senescence and metabolic pathways [14, 20, 21]. Moreover, WRKYs members appear to be tightly involved in the response regulation to various biotic and abiotic stresses [20, 22, 23]. The WRKY gene family has been described and studied in several plant species namely arabidopsis [14, 15], rice [24, 25], cucumber [26], barley [18], maize [27], tomato [28], physic nut [29] and cotton [30]; however, little is known about citrus.
Our work aims to identify all putative WRKY TFs in Citrus and thus to propose a list of candidate genes related to disease resistance, fruit acidification, and environmental stresses adaptation. Detailed analyses of WRKY in Citrus including gene classification, multiple alignment, gene phylogeny, conserved motif composition, and hierarchical classification of ESTs have been performed. The recently sequenced sweet orange genome has provided a reference genome for analyses of chromosomal locations, syntenic relationships and the expression of the CsWRKYs in different tissues. Since available data on Citrus WRKY microarray are missing, we performed a study on gene functionality and expression patterns using Genevestigator tools in comparison with the closest homologous from Arabidopsis. We founded that two Citrus WRKY genes (CsWRKY12 and CsWRKY14) may be proposed as candidate genes potentially up-regulated under all abiotic stress conditions. Also, a hypothesis can then be issued on the possible involvement of Cc53, Cc54, Cs30, Cc60, Cc67, Cs40, Cs41 and Cs48 genes in the response of Citrus to drought and salinity.
Our results from the genome wide survey of Citrus WRKYs family provide useful basis for further and targeted molecular analysis and functional characterization of potential WRKY candidate genes.
Methods
Sequence Database Searches
To identify putative WRKY genes in Citrus, two strategies were adopted as follows: firstly, all Arabidopsis thaliana WRKY family members retrieved from Plant TFDB database (http://planttfdb.cbi.edu.cn/) [31] were used as queries to search homology within Citrus genomes using tBLASTn [32]. The e-value threshold was set to 5 × 10−06; however, only one sequence was discovered in NCBI which is CuWRKY1.
Secondly, query search using “WRKY & Citrus” keywords within ten DB: NCBI (http://www.ncbi.nlm.nih.gov), TIGR (http://plantta.jcvi.org/), CGF (http://cgf.ucdavis.edu/home/), CFGP (http://bioinfo.ibmcp.upv.es/genomics/cfgpDB/index.html), CGD (http://www.Citrusgenomedb.org/), Plant TFDB (http://planttfdb_v1.cbi.pku.edu.cn), HarvEST (http://harvest.ucr.edu/), Phytozome (http://www.phytozome.net), ICGC (http://www.Citrusgenome.ucr.edu/) and CBL (http://biotecnologia.centrodecitricultura.br/index.html). Further analyses, based on WRKY domain presence as given by PFAM, revealed 1868 potential WRKY TFgenes. All these sequences were given a designation number from 1 to 1868 depending on Citrus species like CsWRKY for Citrus sinensis, PtWRKY for Poncirus triofiolata, etc.
In order to increase the reliability of our results and avoid redundancy within the identified WRKY genes in Citrus, sequences were filtered using NCBI local Blast standalone (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/README.bls) [33] and then assembled with CAP3 (Sequence Assembly Program) (http://pbil.univ-lyon1.fr/cap3.php/) [34]. We thus proposed a unique WRKY identifier and followed Arabidopsis classification for WRKY genes annotation.
Annotation and Classification of WRKY Family in Citrus
Full-length WRKY sequences obtained were used for further analysis. They were blasted in Genbank database to retrieve A. thaliana WRKY homologous. Filtration of the resulting alignments was performed using a threshold e-value of 10−10 and WRKYs annotation according to Arabidopsis classification (groups I, IIa, IIb, IIc, IId, IIe, and III) was followed. Citrus WRKY domain was aligned using Clustal X 1.83 with default settings [35]. CsWRKY genes were located on chromosomes in Phytozome database (Sweet Orange Genome Project 2010, http:://www.phytozome.net/orange). Besides, CcWRKY genes were located on scaffold in Phytozome database (Haploid Clementine Genome, International Citrus Genome Consortium, 2011, http://int-Citrusgenomics.org/, http:://www.phytozome.net/clementine). All gene structures (exon-intron organization) were predicted by FGENESH Softberry (http://linux1.softberry.com/berry.phtml) [36].
Multiple Sequence Alignment, Structural and Phylogenetic Analysis
To gain insights into the evolutionary relationship between Citrus and Arabidopsis WRKY proteins, multiple alignments, motif study and phylogenetic analysis were performed. All Citrus WRKY proteins and representative members of all subgroups of AtWRKY protein (AtWRKY20 (At4g26640), AtWRKY40 (At1g80840), AtWRKY72 (At5g15130), AtWRKY50 (At5g26170), AtWRKY74 (At5g28650), AtWRKY65 (At1g29280) and AtWRKY54 (At2g40750)) were sequences aligned by Clustal X 1.83 using default settings [35]. Phylogenetic studies of the WRKY proteins family were conducted using MEGA version 4 [37], Jones-Taylor Thornton (JTT) and Neighbor-Joining (bootstrap = 1000). The evolutionary distance is counted using this method [38].
Multiple Expectation Maximization for Motif Elicitation (MEME) program was accessed online (http://meme.sdsc.edu/meme/intro.html) [39] and used to highlight structural variations among Citrus WRKY proteins. The parameters were as followings: number of repetition—any, maximum number of motifs = 10, and the optimum motif width was maintained to between 6 and 50 residues.
Chromosomal Locations, Syntenic Relationships and Protein Properties of WRKY in Sweet Orange
In order to determine the chromosomal location of CsWRKY2 to CsWRKY52, BLASTn was performed on Orange Genome [40] (citrus.hzau.edu.cn/orange).
MapChart 2.2 [41] was used to visualize chromosome linkage on the ten citrus chromosomes. The alignment of genes coding sequences within individual duplicate blocks was realized by Clustal W software. The Pamilo-Bainchi-Li substitution model, the bootstrap variance estimation method (1000 replicates) and the pairwise deletion were selected when performing codon-based Z test for each block [42, 43]; and only those showing significant results (P value <0.05) were considered.
The iTAK database (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi) currently contains the identified TFs from sweet orange with fully sequenced genomes [44]. Synteny analysis was performed using Plant Genome Duplication Database [45]. Syntenic blocks were drawn using Circos Tool.
Physico-chemical properties, molecular weight and isoelectric point of CsWRKYs were determined on ProtParam tools website (http://web.expasy.org/protparam/) [46].
Hierarchical Classification of Citrus WRKY ESTs into Expression Groups
For cluster patterns expression analysis of WRKY ESTs, HarvEST Citrus database (http://www.harvest-web.org/) was used in order to examine the distribution of 132 Citrus-WRKY unigenes present into the 90 cDNA libraries, with a total number of ESTs of 985. The presence/absence of WRKY ESTs in each cDNA libraries was recorded as (0 or 1) matrix and used to construct a simple model distance matrix and hierarchical dendrogram using the method of Ward [47] with appropriate functions (“dist” and “hclust”) in R language [48].
Expression Patterns of CsWRKY Genes
RNA-seq data provided by Xu et al. [40] from the sweet orange genome database was employed to retrieve fragments per kilobase of transcript per million fragments mapped values (FPKM). The Multiple Array Viewer MeV v4.4.1 [49] was used to cluster the expression values of the 51 CsWRKY genes.
Plant Expression Database Exploitation for WRKY Gene Expression Studies in Citrus
For WRKY gene expression analysis in citrus, publicly available microarray data were obtained and consulted from Plant Expression Database (PLEXdb, http://www.plexdb.org/) [50]. BLASTN served to identify WRKY genes probe sets (e-value, 4.00E−04). We could thus follow expression profiles of WRKY genes under several treatments of stress and different culture conditions. The GeneChip® Citrus Genome Array obtained from PLEXdb allowed us to generate expression data from seven platforms containing 69 hybridizations and from which raw data from every experimental conditions were normalized for further studies. As previously done, the Multiple Array Viewer MeV v4.4.1 [49] was also used to cluster the expression values.
Analysis of Expression Profiles Under Biotic and Abiotic Stresses
Public expression data making available multiple datasets from several experiments and conditions such as Genevestigator (www.genevestigator.ethz.ch) are expanding [51, 52]. Given that no microarrays data were available in Citrus, we used Genevestigator data with the nearest homologs in Arabidopsis, assuming that protein function is conserved between orthologous sequences. Genevestigator is capable of performing large assays on thousands of microarrays giving repeatable and reliable results [51, 52]. We looked for the nearest homologous to Arabidopsis of the 100 full-length WRKY genes. We presented developmental and tissue-specific gene expressions of 20 AtWRKY genes found in Genevestigator and possibly involved in (a)biotic stress tolerance. We chose the hierarchical clustering tool to identify groups of genes that have similar expression profiles. Log2 values for the expression of 20 genes in various stress profiles and following the application of certain stimuli were imported from Genevestigator. The hierarchical classification of 20 different transcriptomes revealed different AtWRKY expressions at different conditions of stress profiles.
To assess the WRKY co-expression network under stress conditions, Genevestigator “co-expression” tool was selected to calculate the coefficient of Pearson correlation particularly among genes proposed as candidates. Thus, a network could be drawn to show the strongly (negatively or positively) correlated genes which have a Pearson Correlation Coefficient PCC value outside the range of [−0.6, 0.6].
Results
Identification of WRKY Family Members in Citrus
As mentioned in “Methods,” several public databases were used to identify WRKY family genes in citrus. The homology search with AtWRKY representative members by tBLASTn provided one single gene from Citrus unshiu (AB573149.1). The keyword search within the ten DB gave a total number of 1868 putative WRKY genes (1768 partial and 100 full-length sequences) belonging to 19 different citrus species (Additional file 1), C. sinensis (sweet orange) being the most represented species with a frequency of 38 %. Similarly, the two Citrus clementina (15 %) and Citrus reticulata (26 %) species are strongly present in most DB. In fact, C. sinensis and C. clementina are part of the sequencing project of Citrus genomes [4], whereas C. reticulata is of commercial market interest.
Thus, 99 full-length sequences were identified within the Phytozome DB and the last and only one of C. unshiu was retrieved from NCBI. All other databases (TIGR, CGF, CFGP, CGD, PlantTFDB, HarvEST, ICGC, and CBL) gave only EST type sequences of which 1343 were from HarvEST (Additional file 1). In order to determine the accurate and non-redundant number of WRKY genes, the local alignment by BLAST at the NCBI allowed the reduction of the number of sequences from 1868 to 1395 and the program CAP3 allowed the assembly of the various EST to form contigs and reduce the number of sequences to a total of 552.
Classification and Molecular Characterization of Citrus WRKY Proteins
A set of 100 full-length sequences of WRKY genes belonging to three Citrus species were identified: 1 in C. unshiu, 51 in C. sinensis (CsWRKY2 to CsWRKY52), and 48 in C. clementina (CcWRKY23 to CcWRKY71) (Additional file 2). Based on the AtWRKYs classification and WRKY domain alignment of these 100 WRKYs by Clustal X 1.83 [35] (Fig. 1), three main groups could emerge. Seventeen members harboring two WRKY domains were assigned to group I for whose the zinc finger motif was C2H2 type [C-X4-C-X22–23-H-X1-H]. Seventy members containing only one WRKY domain, of which the zinc finger motif is C2H2 type in the form of [C–X4–5–C–X23–H–X1–H], were assigned to group II including the five following subgroups: group II-a (5 members), group II-b (21 members), group II-c (23 members), group II-d (10 members), and group II-e (11 members). The group III comprises 13 members with also only one WRKY domain and where the zinc finger motif was C2HC [C–X7–C–X23–24–H–X1–C] or [C–X4–C–X23–H–X1–C] type (Additional file 2).


Multiple sequences alignment of the WRKY domains from Citrus with selected Arabidopsis strains WRKY transcription factors. The highly conserved WRKYGQK and the zinc finger motif sequences are highlighted in yellow, while the mismatched amino acids are marked in blue. Gaps are marked as dashes
Several WRKYs have been described and characterized in other plant species namely Arabidopsis (74 AtWRKYs) [15], rice (102 OsWRKYs) [25], poplar (104 PtWRKYs) [53], ricin (47 RcWRKYs) [54], cucumber (55 CsWRKYs) [26], tomato (81 SlWRKYs) [28], barley (45 HvWRKYs) [18], maize (136 ZmWRKYs) [27] and cotton (112 GrWRKYs and 109 GaWRKYs) [55]. The different WRKY subgroups from C. sinensis, C. clementina, A. thaliana, Vitis vinifera, Oryza sativa and Cucumis sativus are described in Table 1. In our work, 51 WRKY sequences have been identified in C. sinensis, 48 WRKY sequences in C. clementina and a single sequence in C. unshiu. In comparison with Arabidopsis (125 Mb), rice (480 Mb) and cucumber (376 Mb) genomes, Citrus genome’s size is quite average with 372 Mb and the number of WRKY sequences is approximately close to the other species. Monocotyledonous plants display a higher number of WRKY proteins than dicots; indeed, maize and rice have respectively 136 and 109 WRKY proteins, while Arabidopsis has only 71.
The length of Citrus WRKYs ORF ranged from 321 bp (CsWRKY48) to 2166 bp (CsWRKY2), encoding polypeptides of 106 to 721 residues. The predicted molecular weight ranged from 12.7 kDa (CsWRKY48) to 77.8 kDa (CsWRKY2), with estimated isoelectric points from 4.7 (CsWRKY45) to 10.1 (CsWRKY48). Serine, a polar amino acid that may participate in hydrogen bonds, was the most abundant residue in 96 % of the citrus WRKY proteins. Besides, the number of exons of the WRKY sequences ranged from 2 to 7, with an average of 3 exons. A noteworthy intronic sequence was found within all WRKY domains found in the Citrus-WRKY genes; however, the N-terminal WRKY domain of the group I was free from intron. Even the WRKYGQK sequence was defined by Eulgem et al. [15] as the conserved heptapeptide characterizing WRKY proteins, for some species like rice, it was observed slight variations in this sequence [56, 57]. Similarly, some Citrus WRKYs displayed substitutions in their WRKY signature. In fact, the CcWRKY59, CcWRKY62, CsWRKY39 and CsWRKY42 sequences present the WRKYGKK domain. Furthermore, CsWRKY43 and CcWRKY63 sequences possess the WKKYGQK signature. One can assume that the variation of the WRKYGQK signature could affect its binding affinity to the W-box and thus the protein function and activity.
Multiple Alignments and Phylogenetic Analysis Based on the WRKY Domain
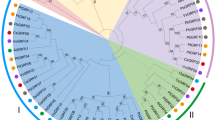
Multiple alignments of amino acid residues of both CsWRKY and CcWRKY showed great divergence within each other respectively. Among the 48 CcWRKYs and the 51 CsWRKYs, both CcWRKY27 with CcWRKY30 and CsWRKY7 with CsWRKY9 showed 55 % sequences identities. Overall, identity between Citrus WRKYs ranged from 30 to 55 % indicating low degree of conservation. In order to examine the phylogenetic relationship among the 100 Citrus WRKYs, the phylogenetic tree was constructed. Sequences from Arabidopsis WRKY members were included in our analysis as references. An unrooted phylogenetic tree was constructed depending on alignments of amino acid sequences of 166 WRKY domains from the four species: A. thaliana, C. clementina, C. sinensis, and C unshiu. As shown in Fig. 2, several subgroups were clearly formed on the basis of the phylogenetic analysis. Even phylogenetic trees of WRKY proteins usually display monophyletic nature [58], in our case, the attribution to defined subgroups is polyphyletic, according to the number of WRKY domains and the type of zinc finger motif. Additionally, the phylogenetic tree of WRKY domains from these species displayed a scattered distribution in all the groups indicating that the expansions of WRKY genes occurred before the divergence of Citrus and Arabidopsis. It has been reported that members of WRKYI group are the most ancient, with loss and/or gain of the N-terminal WRKY domain during the WRKY gene evolution process, and those of groups II and III are thought to be derived descendants from WRKYI group [19]. The groups IIb and IIc are divided into other groupings according to the nearest homologous with Arabidopsis. Subgroup IIc is closely linked to the group I with the exception of CsWRKY52 and CcWRKY70 and the subgroup IIb is bound to the subgroup IIa. This separation in subclades of the WRKYII group domains highlights their paraphyletic nature. On the other hand, in Fig. 2, the tree shows that group III is monophyletic.

Unrooted phylogenetic tree representing relationships among Citrus and selected Arabidopsis WRKY protein. The phylogenetic tree from amino acid sequences was depicted by the MEGA v4.0 program with the neighbor-joining method. Clades of the Citrus WRKY domain are labeled according to the classifications of AtWRKY domains by Wu et al. [24]
As these WRKYs are involved in plants adaptation to various types of environmental stresses, duplication phenomenon occurrence within their members would contribute to the raise in acclimatization ability and rapid implementation of different signal transduction cascade of stress [27]. Based on comparative analysis among all kinds of WRKY genes plants, Zhang and Wang [19] have described an expansion of WRKY members in a more complex way towards pluricellular species.
Conserved Motifs and Domains in Citrus WRKY Proteins
We confirmed for all the identified sequences the presence of the WRKY conserved domain referenced as PF03106 WRKY DNA binding domain [15] and annotated by the followings terms Gene Ontology: 0003700, “ DNA binding Transcription Factor Activity” and Gene Ontology: 0006355, “ transcriptional control” [58]. Except some sequences, Citrus WRKYs proteins showed the PF10533 “Zinc Plant Cluster domain.”
Using MEME software, motif distribution and domain prediction could be assessed (Fig. 3). The first and second motif correspond to WRKY domains, which were broadly distributed in the Citrus WRKY protein sequences. The third motif is present in all groups except for the groups IIa, IId, III and some sequences like CcWRKY63 and CcWRKY64. This pattern is generally located upstream of WRKY domain. The motifs 4 and 8 are present only in group I: pattern 4 corresponds to the second conserved domain WRKY of group I while pattern 8 corresponds to the zinc finger motif. The patterns localization of each Citrus WRKY sequence is close to each other. In addition, there are several patterns (motifs 1, 2, 3, 5, 6, 9, 10) in the subgroup IIb. Some motifs like 5, 10 and conserved NLS motifs were mainly found in subgroups IIa and IIb, while others like motif 6, whose function remains unknown, were only located in subgroups IIa and IIb. Motif 7, a calmodulin-binding conserved domain (CaMBD), is often present in subgroup IId,, indicating putative involvement in signal transduction via activity regulation of target proteins [59]. Additionally, this CaMBD motif was found in CcWRKY33 and CsWRKY13 of the subgroup IIe. Besides, several motifs could be likewise revealed by MEME, yet with uncharacterized function. This may indicate a high and subtle regulation of WRKY biological roles from Citrus.

Ten predicted sequence motifs were identified by the Pfam database and analyzed by Meme V4.8.1. Different motifs, numbered 1–10, are displayed in different colored boxes. The conserved motifs 1, 2, 3, 5, and 11 were definitely identified as WRKY domains, which are broadly distributed in the Citrus WRKY family. The names of all members and subgroups are displayed on the left-hand side, while the exact length of motif is shown at bottom of the figure
Chromosomal Position and Synteny Analysis of the CsWRKY Genes
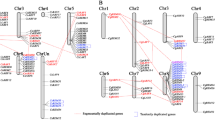
In silico assessment of genes chromosomal location revealed a sparse distribution of the 51 CsWRKY genes across all ten sweet orange chromosomes (Table 2, Fig. 4). Chromosome VII harbored 11 WRKY genes, the highest number of WRKY genes located on the same chromosome. The uncharacterized and II chromosomes contained, respectively, nine and seven WRKY genes. In contrast, chromosomes III and VIII encompassed each only one WRKY gene. Besides, five gene pairs are suspected to be segmental duplicated among the sweet orange chromosomes (Fig. 4) presumably resulting from the polyploidy event in this genome: CsWRKY12 and CsWRKY2, CsWRKY14 and CsWRKY2, CsWRKY4 and CsWRKY46, CsWRKY40 and CsWRKY48, CsWRKY41 and CsWRKY48, CsWRKY27 and CsWRKY18; assuming thus functional divergence between duplicated genes. Each of those genes pairs belongs to the same group of WRKY. Moreover, the phylogenetic analysis performed by MEGA 6 software (Additional file 3) let assume very close relationships among CsWRKYs.

Localization of the identified CsWRKY genes on the sweet orange chromosomes. The chromosome number is indicated at the top of each chromosome. Identical colored circles or squares indicate potential segmental duplicated WRKY gene pairs. The genes marked in different color indicate the group of the WRKY family (I, pistachio green; IIa, burgundy; IIb, blue; IIc, green; IId, blue sky; IIe, pink; III, red)
An extensive number of Arabidopsis model plant WRKYs have been widely investigated [15, 60] and thus we performed for Citrus WRKYs an analysis of synteny (Fig. 5) that led to the identification of 17 pairs of syntenic relations.

Chromosome distribution and synteny analysis of sweet orange WRKY genes. Chromosomes 1–Unch are shown with different colors and in a circular form. The approximate distribution of each CsWRKY gene is marked with a short red line on the circle. Colored curves denote the details of syntenic regions between CsWRKY genes
Several citrus orthologs namely CsWRKY11, CsWRKY27, and CsWRKY33 displayed synteny location with CsWRKY5 and CsWRKY2, CsWRKY 24 and CsWRKY18, respectively.
All these results together suggest a tight conservation of WRKYs family and therefore its involvement and crucial role in plant biology and development.
Hierarchical Classification of WRKY ESTs into Expression Groups
In order to elucidate the possible role of the WRKYs in citrus adaptation to stressful conditions, the expression profile of WRKYs was analyzed under several stimuli such as biotic or abiotic stresses [61] or hormonal treatments [62]. To identify Citrus WRKY ESTs putatively associated with important physiological mechanisms, expression data of 132 unigenes were clustered. It would become thus possible to distinguish through several kinds of treatments diverse and specific patterns of gene expressions. Citrus unigenes can be separated into five main groups based on their library distribution (Fig. 6). The first cluster (biotic stress) grouped unigenes only from UCRPT01 library that displays induced WRKY ESTs in response to Tristeza Virus infection, the most damaging virus affecting Citrus [63]. The second cluster (no treatment) containing ESTs from various libraries is divided into two subclusters: the first, grouped unigenes only from CitNFL library which concern mixed tissue of C. reticulata and the second grouped unigenes from various libraries. The third cluster contains unigenes from libraries involved in plant development by ethylene (AbsAov1). The remaining unigenes (group of pathogen) were distributed libraries of UCRCS11 (red scale), CS00-C1-401, and CM30-C1-401 (Citrus leprosis virus CiLV). The biggest and last cluster grouped unigenes from libraries involved mainly in abiotic stress. The most represented library in this cluster were FLAVEDO4 (heat) and UCRCS06 (storage).

Dendrogram showing relationships among Citrus WRKY unigenes based on their expression data
Expressions of Citrus WRKYs in Different Tissues
Transcriptome sequencing data of several citrus tissues and organs (i.e., callus, fruit, flower, and leaf) [40], publicly available, were surveyed and exploited in order to retrieve and study expression profiles variations of CsWRKY genes (Table 2), and FPKM value was employed to determine the levels of genes expression. Thirty-eight genes at least of the 51 checked CsWRKY were significantly induced in one of the four organs. All the top five highly expressed CsWRKY genes, including CsWRKY28, CsWRKY39, CsWRKY25, CsWRKY14 and CsWRKY12, were detected in callus, suggesting a probable role in meristematic cell growth and development.
A constitutive expression was reported in all tissues and organs for CsWRKY2, CsWRKY7, CsWRKY9, CsWRKY18, CsWRKY19, CsWRKY23, CsWRKY26, CsWRKY28 and CsWRKY32 (Fig. 7), callus being the tissue displaying the highest level of gene expression. Meanwhile, CsWRKY52 is down-regulated in all the organs.

Expression analysis and hierarchical clustering of all WRKY genes in C. sinensis in different organs (leaf, callus, flower, fruit). Heat map representation and hierarchical clustering of the CsWRKY genes. The FPKM values retrieved from published RNA-seq data were log2 transformed and the heat map generated using TIGR MeV v4.1.1. Color scale at the end of the dendrogram represents relative expression levels: green represents low level and red indicates high level; asterisk indicates WRKY genes
Callus displayed genes exclusively up-regulated in this tissue, namely CsWRKY8, CsWRKY10, CsWRKY22, CsWRKY24, CsWRKY31, CsWRKY33, CsWRKY40, CsWRKY49 and CsWRKY51. CsWRKY15 and CsWRKY41 were both expressed in fruit, flower, and leaf, and CsWRKY38 was specifically and solely transcribed in flower.
The transcriptional analysis of 31 CsWRKY and 28 CcWRKY genes within citrus organs and fruit ripening stages showed variations in expression patterns, indicating their putative role in fruit maturation. Particularly, a transcriptional analysis of epithelial cells in grapefruit pee, where it is assumed that secretory glands synthesize citrus essential oil, showed that CsWRKY10–51 and CcWRKY34–58 were up-regulated in comparison with the parenchyma cells, the non-oil-biosynthesizing cells, as the control cell type. These groups of WRKY genes highly expressed in epithelial cells could be involved in the essential oil biosynthetic pathway.
As citrus pulps contain a wide range of acidity content depending on genotypes, we made a comparison at the transcriptional level between three lemon genotypes differing in their acidity (standard, sweet, and acid). We found that CcWRKY34–62 and CsWRKY42–10 displayed higher transcript levels in acidic fruits than sweetness fruits, indicating their possible role in fruit acidification during ripening (Fig. 8).

Expression analysis and hierarchical clustering of WRKY genes in C. sinensis and C. clementina (a) and under different conditions: CT1 (Lemon_acidity) and CT10 (Transcriptional analysis of epithelial cells in grapefruit peel) in C. sinensis (b) under different biotic stress: CT2 (Transcriptional analysis of the sweet orange interaction with the citrus canker pathogens), CT5 (Expression data of sweet orange in response to Candidatus Liberibacter asiaticus infection at advanced stage), CT7 (Gene expression in C. sinensis following infection with the bacterial pathogen Candidatus Liberibacter asiaticus causing Huanglongbing in Florida), and CT11 (Global changes in gene expression of grapefruit peel tissue in response to the yeast biocontrol agent Monilinia fructicola) in citrus sinensis (c) under different conditions: CT1 (Lemon_acidity) and CT10 (Transcriptional analysis of epithelial cells in grapefruit peel) and CT8 (Comparative analysis of stylar canal cells transcriptome in clementine mandarin) in C. clementina. Color scale at the end of each dendrogram represents relative expression levels: blue represents low level and yellow indicates high level; asterisk indicates WRKY genes
Microarray data (CT8) related to self-incompatibility in clementine did not reveal any interesting candidate for this biological mechanism (Additional file 4).
Transcription factors (i.e., WRKYs) whose expression is elevated in particular tissues regulate in all likelihood transcription activity of genes involved in plant growth and developmental processes [64–66].
In Silico Analysis of Expression Profiles Under Abiotic Stress
A preliminary way to elucidate genes function is to analyze their spatio-temporal expression levels. Abiotic and biotic stresses are known to induce the expression of a multitude of WRKYs [67], meaning their role in regulatory transcription activity under such constraints. Some AtWRKYs (Additional file 3) were selected for the study of their expression under abiotic stress within five different tissues: root culture, seedling, rosette, adult leaf, and roots. In normal conditions, almost all WRKY genes are expressed especially in roots, where perception of various environmental stresses is proceeded.
In contrast, some genes such as AtWRKY70 reveal a difference in expression in adult leaf. Indeed, the root system is an organ of perception of various environmental stresses.
To define the specificity and the interaction between the transcriptomic responses to the different treatments studied, we analyzed the over-expressed and under-expressed genes. The data set shows that the same TF can be induced or repressed under different stress states (Fig. 9). Our data matrix included 44 different abiotic stresses (anoxia, cold, drought, dark, heat, hypoxia, etc.). CsWRKY12 and CsWRKY14 displayed high levels of transcripts under all stress conditions and thus may be proposed as candidate genes potentially involved in abiotic stress response, particularly drought and heat adaptation. Moreover, these latter were found to be co-expressed with a salt inducible zinc finger gene (Fig. 9e) in a highly correlated manner, confirming thus their involvement in abiotic stress tolerance.



Profile of WRKY gene expression under abiotic stress by the Genevestigator database. a Percentage of potential WRKY gene expression in different tissues of A. thaliana. b Hierarchical clustering of 20 AtWRKY under various abiotic stress conditions in A. thaliana (log 2 scale). The genes up-regulated are marked in red, while the down-regulated genes are marked in green. Summary of the expression analyses of WRKY genes up-regulated (c) and down-regulated (d). e The dendrogram indicates the degree of similarity (Pearson’s correlation) between gene expression profiles and WRKY conditions tested. Pearson Correlation Coefficient of AtWRKY33 (potentially CsWRKY12 and CsWRKY14)
In our bicluster data matrix, nine AtWRKY genes (W33, W18, W40, W75, W6, W48, W28, W11, W22 and W46) were expressed as specific mediators in salt response (150 mM NaCl). For example, WRKY33 (At2g38470), the nearest homologous to CcWRKY52 and CsWRKY29, was induced in roots treated by NaCl [68], which corroborates their mediation in salinity adaptation. The pathway analysis shows that induction of WRKY33 expression depends in part on the Abscisic acid (ABA), but is independent of the SOS system (Salt Overly-Sensitive) [68]. Transgenic plants over-expressing either WRKY25 or WRKY33 better behave under salinity conditions than the sensitive double mutant wrky25-wrky33 [69]. Drought stress which is often linked to salinity generally reduces plant growth and crop productivity [70]. A hypothesis can then be issued on the possible involvement of Cc53, Cc54, Cs30, Cc60, Cc67, Cs40, Cs41 and Cs48 genes in the response of Citrus to drought and salinity.
In our analysis, AtWRKY40 was expressed in response to cold (4 °C). According to the results of Genevestigator, Cc53, Cc54, Cs30, Cs28, Cs32, Cc51 and Cc55 TFs could be potentially implicated in cold stress tolerance. In contrast, the heat stress inhibits the expression of the following genes: Cs7, Cs9, Cs12, Cs14, Cc60, Cc67, Cs40, Cs41, Cs48, Cs20, Cs19, Cc45, Cs24, Cs33, Cc47, Cc40, Cs28, Cs32, Cc51 and Cc55S.
Expression Studies of WRKYs in Response to Biotic Stress
To assess and confirm Citrus WRKY TFs involvement in the response to biotic stress and the regulation of target genes transcription, AtWRKY orthologs were analyzed for their expression profiles under pathogens infestation like Alternaria brassicicola, Botrytis cinerea, Erysiphe orontii, Golovinomyces cichoracearum, Pseudomonas syringae, Phytophthora infestans and Phytophthora parasitica (Fig. 10). For Arabidopsis microarray analysis, root, adult leaf and rosette were selected. Almost all WRKY genes are expressed, especially in roots under biotic stress, except AtWRKY70 which reveals a difference in expression in adult leaf and rosette.
Microarray data analysis showed that globally 47 Citrus WRKY genes were differentially expressed, among them 24 were up-regulated and 23 were down-regulated under the above-mentioned biotic aggression (Fig. 10). Interestingly, five WRKY genes (Cc60, Cc67, Cs40, Cs41 and Cs48) were always up-regulated by these pathogens infections which make them good candidates for stress adaptation improvement. AtWRKY75, which has been shown as a positive regulator of defense against diseases [71], represents the closely related orthologs of Arabidopsis to these latter (Cc60, Cc67, Cs40, Cs41 and Cs48). In our bicluster data matrix, AtWRKY33, AtWRKY40 and AtWRKY75 genes were up-regulated under Pseudomonas syringae, Phytophthora infestans and Phytophthora parasitica contamination. In contrast, these genes were down-regulated in the case of A. brassicicola and G. cichoracearum infestation. For example, the insert mutant AtW33 (Cs12, Cs14 orthologs) has increased sensitivity to the B. cinerea necrotrophic fungi. The susceptibility of the plant is correlated with weak levels of PR-1 transcripts [72]. Some TFs have been known to be associated in a complex interaction with redundant roles, antagonists or distinct in plant-microbe interactions. This has been demonstrated, for example, for AtW18, AtW40 and AtW60 which act as regulators of basal resistance in plants [73]. The most correlated gene with AtWRKY75 is glutathione S transferase which is implicated in oxidative stress (Fig. 10e).



Profiles of WRKY gene expression under biotic stress by the database Genevestigator: a percentage of potential WRKY gene expression in different tissues of A. thaliana. b Hierarchical clustering of 20 AtWRKY under various biotic stress conditions in A. thaliana (log 2 scale). The genes up-regulated are marked in red, while the down-regulated genes are marked in green. Summary of the expression analyses of WRKY genes up-regulated (c) and down-regulated (d). e Pearson correlation coefficient of AtWRKY75 (potentially CcWRKY60, CcWRKY67, CsWRKY40, CsWRKY41 and CsWRKY48)
Citrus GeneChip array related to canker disease (GPL5731) was prospected to survey the transcription patterns of sweet orange WRKYs (CT2) (Fig. 8, Additional file 4). CsWRKY16 was the only one to be up-regulated by these pathogens, suggesting its putative role in canker disease resistance. Similarly, the Affymetrix GeneChip® citrus genome array (CT5) analysis demonstrated that CsWRKY14 could be associated with defense response against Huanglongbing infection (HLB, Yellow Dragon Disease), the most severe disease of citrus.
Expression Analysis of WRKYs in Response to Hormonal Treatments
In our bicluster data matrix, the most important variation of expression after hormonal stimulus is observed after methyl jasmonate (MeJa) treatment. This treatment represses at the same time AtW3, AtW75, AtW6, AtW23, AtW28, AtW22 and AtW70, and induces AtW40 at the root and aerial parts levels (Fig. 11). The Meja, derived from jasmonic acid, is a plant hormone acting as cell regulator in several of aspects of plant biology, such as flowering, rhizogenesis, seed germination, fruit ripening and senescence [74]. So and by homology, these Cs7, Cs9, Cc60, Cc67, Cs40, Cs41, Cs48, Cc49, Cs31, Cs20, Cs19, Cc45, Cs24, Cs33, Cc47, Cc40, Cs28, Cs32, Cc51 and Cc55 genes would be suppressed after treatment with Meja; however, Cc53, Cc54 and CS30 genes are induced by the same treatment (Fig. 11). ABA plays role in several plant developmental and adaptation processes including stomatal closure, seed germination, embryo maturation, flowering, leaf senescence and stress signal transduction [75]. According to the results of the Genevestigator database, Cs12, Cs14, Cc53, Cc54 and Cs30 are potentially involved in the stress response pathway which is ABA dependent (Fig. 11). Jasmonic acid (JA) and salicylic acid (SA) are phytohormones that function as signaling molecules involved in plant responses to abiotic and biotic stresses as well as plant growth and development namely photosynthesis, transpiration, ion uptake and transport and plant defense responses against pathogens [76, 77]. AtW70 (Cs28, Cs32, Cc51 and Cc55 orthologs) modulates interference in signaling pathways between SA and JA [78].

Profiles of WRKY gene expression in different tissues (a) and under various hormonal treatments (b)
Conclusion
The public availability of citrus genomic and transcriptomic data led us to the identification and characterization of 100 WRKY genes from C. sinensis (51), C. clementina (48) and C. unshiu (1). Our approach allowed us to select interesting citrus WRKY members that play crucial role in plant development and stress responses, and whose further functional characterization would lead to a better understanding of their biological impact in pivotal biological pathways. A combined phylogenetic analysis with WRKY orthologous from Arabidopsis showed that the Citrus WRKY superfamily could be classified in seven groups. We noted that AtWRKYs, CsWRKYs and CcWRKYs characterized by similar functions in stress resistance showed an ability to be grouped. Thus, AtWRKY3 and AtWRKY4, contributing to the defense against B. cinerea [79] were clustered together on the phylogenetic tree with CcWRKY34 and CsWRKY7, both belonging to group I. Similarly, AtWRKY72, the representative gene of the group IIb, involved in basal immunity in Arabidopsis [80], clustered together with CsWRKY51 and CcWRKY31 within the phylogenetic tree. In group IId, AtWRKY11 and AtWRKY17, characterized by their involvement in response to pathogens or nematode [81], clustered together with CcWRKY48 and CsWRKY28. The analysis of several Citrus gene expression data demonstrated the putative implication of WRKY groups or lonely genes in diseases resistance (CTV, CiLV, Canker and yellow dragon diseases and miscellaneous pathogens infestations), environmental stress adaptation (salinity, drought, heat and cold) and various plant biological mechanisms (plant development, cell growth, fruit acidification and essential oil biosynthesis) via hormonal mediation (ABA, ethylene, JA and SA). In conclusion, Citrus WRKYs family is potentially involved in several plant developmental processes, abiotic and biotic stress responses, and thus, members of this family could be suitable candidates for further Citrus breeding and improvement program.
References
Talon, M. & Gmitter, Jr. F.G. (2008) Citrus genomics. International Journal of Plant Genomics.1-18.
http://faostat3.fao.org/browse/Q/QC/E. Accessed March 2016.
Krug, C. A. (1943). Chromosomes numbers in the subfamily Aurantioideae with special reference to the genus Citrus. Botanical Gazette, 48, 602–611.
Gmitter, F. G., Jr., Chen, C., Machado, M. A., Alves de Souza, A., Ollitrault, P., Froehlicher, Y., & Shimizu, T. (2012). Citrus genomics. Tree Genetics & Genomes, 8, 611–626.
Dambier, D., Benyahia, H., Pensabene-Bellavia, G., Aka Kacar, Y., Froelicher, Y., Belfalah, Z., Beniken, L., Handaji, N., Printz, B., Morillon, R., Yesiloglu, T., Navarro, L., & Ollitrault, P. (2011). Somatic hybridization for Citrus rootstock breeding: an effective tool to solve some important issues of the Mediterranean Citrus industry. Plant Cells Reports, 30, 883–900.
Olivares-Fuster, O., Duran-Vila, N., & Navarro, L. (2005). Electrochemical protoplast fusion in Citrus. Plant Cell Reports, 24, 112–119.
ICGC (2003). Draft of the International Citrus Genome Consortium White Paper, 42. Available from http://www.Citrusgenome.ucr.edu/. Accessed November 2012.
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., Putnam, N., Ralph, S., Rombauts, S., Salamov, A., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr & Gray). Science, 313, 1596–1604.
Forment, J., Gadea, J., Huerta, L., Abizanda, L., Agusti, J., Alamar, J., Alos, E., Andres, F., Arribas, R., Beltran, J. P., et al. (2005). Development of a Citrus genome-wide EST collection and cDNA microarray as resources for genomic studies. Plant Molecular Biology, 57, 375.
Terol, J., Conesa, A., Colmenero, J. S., Cercos, M., Tadeo, F., Agustí, J., Alós, E., Andres, F., Soler, G., Brumos, J., et al. (2007). Analysis of 13000 unique Citrus clusters associated with fruit quality, production and salinity tolerance. BMC Genomics, 8, 31.
Bausher, M., Shatters, R., Chaparro, J., Dang, P., Hunter, W., & Niedz, R. (2003). An expressed sequence tag (EST) set from Citrus sinensis L. Osbeck whole seedlings and the implications of further perennial source investigations. Plant Science, 165, 415–422.
Shimada, T., Fujii, H., Endo, T., Yazaki, J., Kishimoto, N., Shimbo, K., Kikuchi, S., & Omura, M. (2005). Toward comprehensive expression profiling by microarray analysis in Citrus: monitoring the expression profiles of 2213 genes during fruit development. Plant Science, 168, 1383.
Rios, G., Naranjo, M. A., Iglesias, D., Ruiz, O., & Talon, M. (2009). Genomics meets induced mutations in Citrus: identification of deleted genes through comparative genomic hybridization. In Q. Y. Shu (Ed.), Induced Plant Mutations in the Genomics Era (pp. 407–410). Rome: Food and Agriculture Organization of the United Nations.
Ülker, B., & Somssich, I. E. (2004). WRKY transcription factors: from DNA binding towards biological function. Current Opinion in Plant Biology, 7, 491–498.
Eulgem, T., Rushton, P. J., Robatzek, S., & Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends in Plant Science, 5, 199.
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C. Z., Keddie, J., Adam, L., Pineda, O., Ratcliffe, O. J., Samaha, R. R., Creelman, R., Pilgrim, M., Broun, P., Zhang, J. Z., Ghandehari, D., Sherman, B. K., & Yu, G. L. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science, 290, 2105–2110.
Rushton, P. J., Macdonald, H., Huttly, A. K., Lazarus, C. M., & Hooley, R. (1995). Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of -Amy2 genes. Plant Molecular Biology, 29, 691–702.
Mangelsen, E., Kilian, J., Berendzen, K. W., Kolukisaoglu, U. H., Harter, K., Jansson, C., & Wanke, D. (2008). Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genomics, 9, 194.
Zhang, Y., & Wang, L. (2005). The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evolutionary Biology, 5, 1.
Rushton, P. J., Somssich, I. E., Ringler, P., & Shen, C. J. (2010). WRKY transcription factors. Trends in Plant Science, 15, 247–258.
Mao, G., Meng, X., Liu, Y., Zheng, Z., Chen, Z., & Zhang, S. (2011). Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell, 23(4), 1639–1653.
Eulgem, T., & Somssich, I. E. (2007). Networks of WRKY transcription factors in defense signaling. Current Opinion in Plant Biology, 10(366–371), 2007.
Pandey, S. P., & Somssich, I. E. (2009). The role of WRKY transcription factors in plant immunity. Plant Physiology, 150, 1648–1655.
Wu, K. L., Guo, Z. J., Wang, H. H., & Li, J. (2005). The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Research, 12, 9–26.
Ross, C. A., Liu, Y., & Shen, Q. J. (2007). The WRKY gene family in rice (Oryza sativa). Journal of Integrative Plant Biology, 49, 827–842.
Ling, J., Jiang, W., Zhang, Y., Yu, H., Mao, Z., Gu, X., Huang, S., & Xie, B. (2011). Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics, 12, 471.
Wei, K. F., Chen, J., Chen, Y. F., Wu, L. J., & Xie, D. X. (2012). Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Research, 19(2), 153–164.
Huang, S., Gao, Y., Liu, J., Peng, X., Niu, X., Fei, Z., Cao, S., & Liu, Y. (2012). Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Molecular Genetics and Genomics, 287(6), 495–513.
Xiong, W., Xu, X., Zhang, L., Wu, P., Chen, Y., Li, M., Jiang, H., & Wu, G. (2013). Genome-wide analysis of the WRKY gene family in physic nut (Jatropha curcas L.). Gene, 524, 124–132.
Dou, L., Zhang, X., Pang, C., Song, M., Wei, H., Fan, S., & Yu, S. (2014). Genome-wide analysis of the WRKY gene family in cotton. Molecular Genetics and Genomics, 289, 1103–1121.
Guo, A. Y., Chen, X., Gao, G., Zhang, H., Zhu, Q. H., Liu, X. C., Zhong, Y. F., Gu, X., He, K., & Luo, J. (2008). PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Research, 36, D966–D969.
Stephen, F., Altschul, L., Madden, T., Alejandro, A., Schäffer, Jinghui, Z., Zheng, Z., Webb, M., & David, J. L. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST +: architecture and applications Software. BMC Bioinformatics, 10, 421.
Huang, X., & Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Research, 9, 868–877.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., & Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23(21), 2947–2948.
Solovyev, V., Kosarev, P., Seledsov, I., & Vorobyev, D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biology, 7, 1–10.
Tamura, K., Dudley, J., Nei, M., & Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and. Evolution, 24(1596–1599), 2007.
Jones, D. T., Taylor, W. R., & Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences, 8, 275–282.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., Ren, J., Li, W. W., & Noble, W. S. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research, 37, W202–W208.
Xu, Q., Chen, L. L., Ruan, X., Chen, D., Zhu, A., et al. (2012). The draft genome of sweet orange (Citrus sinensis). Nature Genetics, 45, 59–66.
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. Journal of Heredity, 93, 77–78.
Li, W. H. (1993). Unbiased estimation of the rates of synonymous and non synonymous substitution. Journal of Molecular Evolution, 36, 96–99.
Pamilo, P., & Bianchi, N. O. (1993). Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Molecular Biology and Evolution, 10, 271–281.
Zheng, Y., & Fei, Z. (2014) iTAK—plant transcription factor & protein kinase identifier and classifier. Fei Bioinformatics Lab, Boyce Thompson Institute and USDA Robert W. Holley Center.
Lee, T. H., Tang, H., Wang, X., & Paterson, A. H. (2013). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Research, 41, D1152–D1158.
Gasteiger; E., Hoogland, C., Gattiker; A., Duvaud, S., Wilkins, M.R., Appel; R.D., & Bairoch, A. (2005) Protein identification and analysis tools on the ExPASy server;(In) John M. Walker (ed): The proteomics protocols handbook, Humana Press. pp. 571–607.
Becker, R. A., Chambers, J. M., & Wilks, A. R. (1988). The New S Language: a programming environment for data analysis and graphics. Pacific Grove: Wadsworth & Brooks/Cole. ISBN 0-534-09192-X.
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/.
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., Braisted, J., Klapa, M., Currier, T., Thiagarajan, M., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques, 34, 374–378.
Dash, S., Van Hemert, J., Hong, L., Wise, R. P., & Dickerson, J. A. (2012). PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Research, 40(D1), D1194–D1201.
Zimmermann, P., Hirsch-Hoffmann, M., Hennig, L., & Gruissem, W. (2004). GENEVESTIGATOR Arabidopsis microarray database and analysis toolbox. Plant Physiology, 136, 2621–2632.
Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., Scholkopf, B., Weigel, D., & Lohmann, J. U. (2005). A gene expression map of Arabidopsis thaliana development. Nature Genetics, 37, 501–506.
He, H., Dong, Q., Shao, Y., Jiang, H., Zhu, S., Cheng, B., & Xiang, Y. (2012). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Reports, 31, 1199–1217.
Li, H.-L., Zhang, L.-B., Guo, D., Li, C.-Z., & Peng, S.-Q. (2012). Identification and expression profiles of the WRKY transcription factor family in Ricinus communis. Gene, 503, 248–253.
Ding, M., Chen, J., Jiang, Y., Lin, L., Cao, Y., Wang, M., Zhang, Y., Rong, J., & Ye, W. (2015). Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Molecular Genetics and Genomics, 290, 151–171.
Zhang, Z. L., Xie, Z., Zou, X., Casaretto, J., Ho, T. H., & Shen, Q. J. (2004). A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiology, 134, 1500–1513.
Xie, Z., Zhang, Z. L., Zou, X. L., Huang, J., Ruas, P., Thompson, D., & Shen, Q. J. (2005). Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiology, 137(1), 176–189.
Babu, M. M., Iyer, L. M., Balaji, S., & Aravind, L. (2006). The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Research, 34(6505–20), 2006.
Park, C. Y., Lee, J. H., Yoo, J. H., Moon, B. C., Choi, M. S., Kang, Y. H., Lee, S. M., Kim, H. S., Kang, K. Y., Chung, W. S., Lim, C. O., & Cho, M. J. (2005). WRKY group IId transcription factors interact with calmodulin. FEBS Letters, 579, 1545–1550.
Dong, J., Chen, C., & Chen, Z. (2003). Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Molecular Biology, 51, 21–37.
Kilian, J., Whitehead, D., Horak, J., Wanke, D., Weinl, S., Batistic, O., D’Angelo, C., Bornberg-Bauer, E., Kudla, J., & Harter, K. (2007). The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant Journal, 50, 347–363.
Goda, H. (2008). The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. The Plant Journal, 55, 526–542.
Febres Vicente, J., Lee, R.F., & Gloria, A. (2007) Genetic transformation of Citrus for pathogen resistance. In I. A. Khan (Ed), Citrus genetics, breeding and biotechnology, chapter 14 (pp 306–327).
Alexandrova, K. S., & Conger, B. V. (2002). Isolation of two somatic embryogenesis-related genes from orchardgrass (Dactylis glomerata). Plant Science, 162, 301–307.
Luo, M., Dennis, E. S., Berger, F., Peacock, W. J., & Chaudhury, A. (2005). MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 102, 17531–17536.
Yu, F., Huaxia, Y., Lu, W., Wu, C., Cao, X., & Guo, X. (2012). GhWRKY15, a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.), is involved in disease resistance and plant development. BMC Plant Biology, 12, 144.
Ryu, H. S., Han, M., Lee, A. K., Cho, J. I., Ryoo, N., Heu, S., Lee, Y. H., Bhoo, S. H., Wang, G. L., Hahn, S. H., & Jeon, J. S. (2006). A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Reports, 25, 836–847.
Jiang, Y. Q., & Deyholos, M. K. (2006). Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biology, 6, 25.
Jiang, Y. Q., & Deyholos, M. K. (2009). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Molecular Biology, 69, 91–105.
Golldack, D., Lüking, I., & Yang, O. (2011). Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Reports, 30, 1383–1391.
Encinas-Villarejo, S., Maldonado, A. M., Amil-Ruiz, F., de los Santos, B., Romero, F., Pliego-Alfaro, F., Muñoz-Blanco, J., & Caballero, J. L. (2009). Evidence for a positive regulatory role of strawberry (Fragaria x ananassa) FaWRKY1 and Arabidopsis AtWRKY75 proteins in resistance. Journal of Experimental Botany, 60, 3043–3065.
Zheng, Z., Qamar, S. A., Chen, Z., & Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant Journal, 48(4), 592–605.
Xu, X., Chen, C., Fan, B., & Chen, Z. (2006). Physical and functional interactions between pathogen induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell, 18, 1310–1326.
Creelman, R.A., & Rao, M.V. (2002) The oxylipin pathway in Arabidopsis. In Somerville, C.R. and Meyerowitz, E.M., (eds), The Arabidopsis book 1:e0012.
Wasilewska, A., Vlad, F., Sirichandra, C., Redko, Y., Jammes, F., Valon, C., Frey, N. F., & Leung, J. (2008). An update on abscisic acid signaling in plants and more. Molecular Plant, 1, 198–217.
Durrant, W. E., & Dong, X. (2004). Systemic acquired resistance. Annual Review of Phytopathology, 42, 185–209.
Seilaniantz Robert, A., Navarro, L., Bari, R., & Jones, J. D. (2007). Pathological hormone imbalances. Current Opinion in Plant Biology, 10(4), 372–379.
Li, J., Brader, G., Kariola, T., & Palva, E. T. (2006). WRKY70 modulates the selection of signalling pathways in plant defense. Plant Journal, 46, 477–491.
Lai, Z., Vinod, K. M., Zheng, Z., Fan, B., & Chen, Z. (2008). Roles of Arabidopsis WRKY3 and WRKY4 transcription factors in plant responses to pathogens. BMC Plant Biology, 8(1), 68.
Bhattarai, K. K., Atamian, H. S., Kaloshian, I., & Eulgem, T. (2010). WRKY72-type transcription factors contribute to basal immunity in tomato and Arabidopsis as well as gene-for-gene resistance mediated by the tomato R gene Mi-1. Plant Journal, 63(2), 229–240.
Journot-Catalino, N., Somssich, I. E., Roby, D., & Kroj, T. (2006). The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell, 18, 3289–3302.
Acknowledgments
Authors would express their sincere gratitude to Sana Louati Elarbi for the proofreading of the paper. This research work was supported by the Ministry of Higher Education and Scientific Research of Tunisia (LR15CBBC02, laboratory grant).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
M. Ayadi and M. Hanana contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Additional File 1
Summary table of the results of various databases according to the species of Citrus (DOCX 17 kb)
Additional File 2
Annotation of complete WRKY gene in Citrus. (A catalog of WRKY proteins in the Citrus) (DOCX 44 kb)
Additional File 3
Phylogenetic tree from amino acid sequences of CsWRKY was depicted by the MEGA V6.06 program with the neighbor-joining method (PDF 14 kb)
Additional File 4
Overview (summary) of experiments used in Plex database (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Ayadi, M., Hanana, M., Kharrat, N. et al. The WRKY Transcription Factor Family in Citrus: Valuable and Useful Candidate Genes for Citrus Breeding. Appl Biochem Biotechnol 180, 516–543 (2016). https://doi.org/10.1007/s12010-016-2114-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-016-2114-8