
Abstract
The thermal, dynamic, and structural properties of wheat starch–water systems with different levels of water content (11, 35, 40, 42, 45, and 50%, wet basis) were investigated. 1H time domain nuclear magnetic resonance (TD-NMR) spectroscopy was used to interpret and quantify the water transfer and starch transformations in terms of water uptake, granule swelling, amylose leaching, and melting of starch polymers in relation to the different levels of water content. Complementary differential scanning calorimetry (DSC) experiments were performed to study the effects of water content on the degree of starch gelatinization. In particular, this twofold approach was applied to the first endotherm to study the mechanisms of gelatinization with a common heating range both in NMR and DSC. It was shown that the trend of the enthalpy changes in the first phase transition in starch–water (SW) mixtures was strongly correlated with the loss of solid content measured by NMR in the corresponding temperature range (55–70 °C). Based on the evolution of the relative amplitudes of T 2, structural transformations of starch were shown to occur in both crystalline and amorphous regions within SW samples, supporting the fact that the amorphous phase of starch also plays a significant role in the phase transition of granules during gelatinization. This dynamic and hydrothermal approach provided the first NMR-based interpretation of the first endotherm measured by DSC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The interactions between water and starch are of considerable scientific and commercial interest, due to the role of water in the stability and functional properties of the main constituents of the majority of food and pharmaceutical systems. In the native form of starch, the two main polysaccharides, amylopectin and amylose, are organized in granules as alternating semi-crystalline and amorphous layers that form growth rings (French 1972). The amorphous layers are constituted of amylose and non-ordered amylopectin branches, while semi-crystalline regions mainly comprise ordered regions of double helices formed by short amylopectin branches and amorphous regions (Godet et al. 1995; Biliaderis 1992). However, there is no apparent boundary between the amorphous and crystalline domains, and molecules are able to pass from one phase to another. Moreover, the crystallinity in native starch, reported in the literature, is exclusively associated with the amylopectin component (Ratnayake and Jackson 2008) due to ordered crystalline arrangements, i.e., clustering of double helices within A and B crystalline types which trap 8 and 36 water molecules per cell unit, respectively (Wild and Blanshard 1986; Hizukuri 1986). Besides the water within starch crystals, starch powder also contains about 10–20% more mobile water that may be absorbed by the granules and adsorbed on the surface. However, the main water causing the swelling of starch granules is related to hydration when water penetrates first into the amorphous regions. This water absorption must occur in non-crystalline areas since the inter-chain bonds in crystalline areas are too strong and their steric accessibility too difficult for water molecules to be bonded. When absorbed, water establishes hydrogen bonds with the –OH groups of glucose units of starch polymers and acts as an effective starch plasticizer, allowing alignment of the crystallites by extension of inter-crystalline amorphous phases (Slade and Levine 1988). Intact starch granules are almost insoluble in cold water and swell only to a limited extent. Starch granule swelling is enhanced with rise in temperature, and the amylose inside the granules leaches out simultaneously, followed by an increase in viscosity (Donald 2004; Singh et al. 2003). Further heating of water-saturated granules leads to irreversible swelling and at the gelatinization temperature they begin to rupture and collapse, resulting in the dispersion of granule fragments and dissolved molecules. The ability of starch to interact with water and to gelatinize is a property used in many industrial applications, as for example, in foods in order to obtain specific textural properties (Considine and Considine 1982) and rheological properties (Xie et al. 2012).
Apart from the structure of starch granules and their ability to form gels with various textural characteristics, less researched and less well understood are the distribution and dynamic state of water within starch granules (Tang et al. 2000). Many authors have used 1H time domain nuclear magnetic resonance (TD-NMR) spectroscopy to analyze the mobility of water and biopolymers in model systems or starchy products such as bread and cakes (Le Grand et al. 2007; Bosmans et al. 2012; Assifaoui et al. 2006; Pojić et al. 2016). Their aims were to understand changes during starch gelatinization related to physical or chemical treatments and different composition, including the influence of different water content (Bogracheva et al. 2001; Da Silva et al. 1996). Research has mainly focused on the spin–spin (T 2) relaxation behavior of non-uniformly distributed water, as a probe of the microstructure within wheat starch–water mixtures (Le Botlan et al. 1998; Choi and Kerr 2003). However, quantitative information on interactions between starch and water, which play a key role in the gelatinization mechanism, is still lacking. The dependence of water compartmentalization on temperature-induced microstructural changes in wheat starch-based model systems and wheat flour dough was recently investigated in great detail and in dynamic experimental conditions (during heating) (Rondeau-Mouro et al. 2015). Differential scanning calorimetry (DSC), which has been proved to be of considerable value in studying thermal transitions, is a complementary technique widely used to characterize starch gelatinization (Bosmans et al. 2016; Schirmer et al. 2015). DSC scanning has shown that when wheat starch granules are suspended in an excess of water (>60%, calculated on wet basis (wb)), all starch crystallites melt cooperatively, exhibiting a single endothermic peak. However, when the amount of water is reduced (<60% wb), a split into two endotherms occurs (Jankowski and Rha 1986). Further water reduction causes the first endotherm to fade and the second endotherm to shift toward a higher region (Svensson and Eliasson 1995). In addition to the DSC studies, two-step wheat starch gelatinization has also been revealed using viscometry measurements (MacArthur and D’appolonia 1979; Medcalf and Gilles 1965). Despite numerous gelatinization models, the molecular mechanism governing the appearance of the two-step gelatinization endotherm is poorly understood. Furthermore, some authors used the complementarity between NMR and DSC to investigate the role of water–starch interactions in the gelatinization of starches from various botanical origins, with or without additives, but the results were discussed separately without explaining the two-step endothermic phenomenon (Tananuwong and Reid 2004; Gonera and Cornillon 2002). Few studies have used both analytical techniques on wheat starch model systems (Chiotelli et al. 2002; Chinachoti et al. 1991; Bosmans et al. 2012). Most interesting were the investigations of Chiotelli et al., who combined DSC and NMR relaxation measurements and proposed expansion of this study over a range of levels of water content and temperature (Chiotelli et al. 2002). This is the subject of the present study in which the final goal was to correlate molecular mechanisms of gelatinization highlighted by TD-NMR with the two-step endotherm observed by DSC. For this purpose, wheat starch–water mixtures with different levels of water content (11, 35, 40, 42, 45, and 50% wb) were heated over a common temperature range and rate between 20 and 90 °C. The main discussion concerns the relative importance of two phenomena: (1) the impact of the water distribution and interactions with starch during starch swelling and gelatinization and (2) the origin of the two-step gelatinization endotherm, tentatively interpreted according to four overriding gelatinization models proposed in the literature.
Materials and Methods
Materials
Native wheat starch (11.00 ± 0.01% moisture wb) was purchased from Sigma–Aldrich (Saint-Quentin-Fallavier, France). In addition to wheat starch powder (SP-11), five wheat starch–water mixtures (SW-35, SW-40, SW-42, SW-45, and SW-50) were prepared by adding deionized water to pre-weighted amounts of starch powder taking into account its own water content. The mixtures were gently homogenized in a glass vial by stirring for 5 min using a small spatula. Each sample was prepared in duplicate (12 samples in total). The dry matter and the water content of all samples were determined by measuring weight difference after drying in an oven at 103 °C for 24 h. Table 1 lists the total water content of the six samples analyzed on wet basis (for a final wet mass of 5 g).
NMR Measurements
NMR measurements were performed with a time domain Bruker spectrometer (The Minispec; Bruker SA, F-67166, Wissembourg, Germany) operating at a resonance frequency of 20 MHz. The 1H NMR probe was regulated by a constant gas flow (compressed air) heated by a variable temperature unit (BVT3000) allowing temperature regulation at ±0.1 °C. Approximately 0.45 g of each sample (starch powder (SP) or starch–water mixtures (SW)) was weighted in 10-mm diameter NMR tubes which were hermetically closed before analysis. A Teflon rod was introduced into the tube to maintain the optic fiber (Neoptix Inc., Canada) which was used for online temperature measurement and to reduce the volume of water vapor. First of all, the relaxation behavior of SW-45 and SW-50 samples was measured and compared at two different heating regimes. The temperature rose between 20 and 90 °C: (1) in a linear modulated temperature ramp using a heating rate of 3 °C/min for 37 min and (2) in step by step heating using 10 °C increments at about 0.2 °C/min for 5 h with 10 min waiting time at the beginning of NMR measurements to assure temperature stabilization and equilibration. In order to monitor changes occurring during heating precisely, T 2 relaxation times were measured at each 10 °C step between 20 and 90 °C in all samples. As in the previous work of Rondeau-Mouro et al. (2015), accurate measurement of T 2 relaxation times could be achieved from combined free induction decay (FID) and the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence (Meiboom and Gill 1958). The probe dead time was 11 μs, and the sampling rate for the FID acquisition was one point per 0.4 μs. The echo time in the CPMG sequence was 0.1 ms using from 500 to 5000 echoes, depending on the temperature and water content of the sample. Sixteen scans were performed for recycle delays varying between 1 and 2.2 s. These delays were measured using the classical inversion recovery (IR) sequence.
Processing NMR Relaxation Data
NMR relaxation curves were analyzed by two methods. The data were fitted using a continuous distribution of T 2 by the Maximum Entropy Method (Mariette et al. 1996), implemented in Scilab (Scilab Enterprises, Versailles, France; license CeCILL, GPL-compatible) and also considering a discrete sum of T 2 by the Marquardt method (Marquardt 1963) based on least squares non-linear regression (Levenberg Marquardt algorithm). All NMR measurements were expressed as spin–spin relaxation times (noted T 2, in ms) with their corresponding relative amplitudes (noted A, in percentages). Careful comparison of results from these two methods revealed that relaxation time values and their corresponding amplitudes were in agreement. The data were also fitted using the Maximum Entropy Method (MEM) on Matlab (The MathWorks, Inc.,Version R2006b 7.3.0.267) to obtain T 2 distributions. Considering the consistency of the results from the MEM and Marquardt methods, only one example of T 2 distributions is presented in appendix for SW-45.
The FID-CPMG data of the starch powder and hydrated starch mixtures were fitted using Eqs. 1 and 2, respectively:
where A(i) and A(j) are the relative amplitudes of the corresponding T 2(i) and T 2(j) spin–spin relaxation times measured during t, the time of the relaxation process. b is a constant, whereas i and j refer to any of the relaxing components in the FID (solid phase) and the CPMG (liquid phase), respectively. The starch powder (SP-11) was characterized by only two T 2 components, whereas the FID-CPMG data of hydrated samples (SW-35 to SW-50) were fitted using the sum of Gaussian and exponential functions depending on the hydration level and temperature of samples.
DSC Measurements
DSC measurements were performed using a Temperature Modulated Differential Scanning Calorimetry Q100 (TM-DSC Q100, TA Instruments) equipped with a cooler system with a 50.0 ml/min flow rate of nitrogen steam. The calorimeter was calibrated in temperature and energy with the standard values of zinc and indium melting points. Approximately 30 mg of sample was weighted in standard stainless steel pans using a microbalance, before they were hermetically sealed. The pans with samples and an empty pan, used as reference, were loaded into the DSC instrument. Programed scanning was carried out from 20 to 130 °C at a heating rate of 3 °C/min. Variations in the enthalpy of phase transition were detected by a difference in heat flow (dQ/dt) between the reference and the sample. All measurements were conducted in at least duplicate and the average values of the thermal parameters were calculated. Following the DSC experiments, samples were reweighted to ensure that DSC pans remained intact after heat uptake and that there was no loss of mass. The real water content of each sample was then determined with a thermogravimetric analyzer (TGA 2050E, TA Instruments, USA) by drying samples at 130 °C for 120 min. All the parameters, such as the onset temperature, T o(P1); two peak temperatures, T p(P1 and P2); conclusion temperature, T c(P2); temperature range in which transition occurred, ∆T (∆T = T c(P2)–T o(P1)); and the enthalpy change undergone by the sample, ∆H, were determined from the DSC thermograms by means of Universal Analysis 2000 software. The enthalpy change was estimated by numerical integration of the area under the thermal transition peak and expressed as joules per gram of dry starch.
Statistical Analysis
All statistical tests were carried out using the software Statgraphics (Centurion XVI) at a significance level of 0.05 (95% confidence interval). One-way ANOVA permitted to detect significant differences (95% degree of confidence) between the NMR parameters (T 2 values and associated signal amplitude) of different model samples. On the other hand, Pearson’s correlation coefficients were calculated to examine the relationship between DSC and NMR data (enthalpy changes during the P1 transition versus the loss of solid content measured by NMR between 55 and 70 °C).
Results and Discussion
Wheat starch–water mixtures with different water content (35, 40, 42, 45, and 50% wb) as well as wheat starch powder (11% wb) were investigated in terms of their relaxation behavior by TD-NMR and their thermokinetic properties by DSC. The first experiments were carried out using the same heating rate (3 °C/min), which is relatively rapid for performing NMR experiments in a good time resolution. Before description of the DSC results and their comparison with those of NMR, T 2 relaxation times measured under the slow heating regime in a stepwise manner (0.2 °C/min) are presented for each sample and interpreted relative to already published results (Bosmans et al. 2012; Rondeau-Mouro et al. 2015; Tang et al. 2000; Tang et al. 2001).
T 2 Relaxation Characteristics Studied by NMR
T 2 relaxation times and their relative amplitudes A for each sample at two temperatures, 20 and 60 °C, are given in Table 2 for various water contents, while an example of T 2 distributions is given for SW-45 in appendix (Fig. 8). As already shown, the relaxation decay curve of the starch powder SP-11 was described by two components, i.e., T 2(1) at 25 μs with an amplitude of 80.4% (±0.1) attributed to protons from macromolecules and T 2(2) at 400 μs representing 19.6% (±0.1) of the total signal that characterized the water protons (Rondeau-Mouro et al. 2015). With addition of water, the relaxation decay curves lengthened. Hydrated starch samples (SW-35 to SW-50) displayed two or three additional components that differed significantly compared to those of starch powder as demonstrated by the ANOVA tests (different group). These differences in T 2 values as well as amplitudes depend on the sample water content and temperature (Table 2). In effect, a fifth component T 2(5) was present in SW-50 throughout the heating program while it appeared at 60 °C and above for SW-40 to SW-45 samples. At short times, the FID signal of hydrated samples (SW-35 to SW-50) was composed of two separate components (T 2(1) and T 2(2)) that were close in values (same homogenous group labeled b) and attributed to protons from the starch macromolecules as already shown for SW-45 (Rondeau-Mouro et al. 2015). T 2(3) showed few variations between samples whatever the temperature and was assigned to intra-granular water protons and represented from 20 to 28% of the total NMR signal at 20 °C, depending on the water content (different homogeneous groups). This component could be distinguished from the extra-granular water by slow diffusional exchange between the two water phases characterized by T 2(4) and T 2(5) components. In fact, the radii of starch granules are in the micrometer range (up to 1–3 μm diameter for spherical A-type, and 20–45 μm diameter for lenticular or disk shaped B-type granules) (Hoseney 1994; Atwell 2001), a distance sufficient to observe the diffusional exchange between water inside and outside the granules. Apparently, the greater the amount of water available in the sample, the higher the relative amplitude of these T 2 components as confirmed by the various group discriminating A(4) at 20 °C for the different samples (ANOVA tests). We can thus assign this last water fraction (T 2 from 7 ms to longer times), characterized by one T 2(4) or two relaxation times T 2(4 + 5), to extra-granular water in high and low interaction with starch, respectively. The mobility of these two extra-granular water phases was analyzed jointly because of their supposed similar composition (greater or lesser starch-rich water phase), differentiated only by the slow diffusional exchange process occurring between them. The fact that at 20 °C, the T 2(4) and T 2(5) relaxation times were centered at about 7.4 to 309 ms (depending on water content in the sample), and not at the bulk water value (∼2.5 s, under the same conditions), indicated that the diffusional water exchange was not in the fast regime on the NMR time scale (Hills et al. 1998).
By heating starch–water samples, significant changes can be expressed for starch and water signal amplitudes. A(1), A(2), and A(5) measured at 60 °C showed indeed significant differences depending on the water content of samples (different groups by the ANOVA tests). Two stages can be envisaged. The first stage (20–50 °C) corresponds to the reversible swelling of starch granules, as a result of the water absorption by starch granules. When starch is suspended in water, the granules absorb water so that the diameter of the granule increases and amylose fragments are leached out, phenomena that are enhanced by the temperature increase. During the second stage (50–90 °C), when the swollen granules are heated further, some hydrogen bonds between adjacent glucose units are disrupted and the crystalline arrangement is partially destroyed. These temperature-associated changes induce the gelatinization of some starch granules, accompanied by damage to their granular structure and higher water–starch interaction. These phenomena were monitored and quantified by the temperature dependence of each T 2 relaxation time and their corresponding relative amplitude (Fig. 1). Evolutions of T 2 relaxation times and relative amplitudes are discussed relative to the signals coming from the FID and CPMG acquisitions. In FID acquisition, random molecular motion of starch constituents characterized by T 2(1) and T 2(2) gave rise to their slight changes (Fig. 1a, b). In contrast to these minor changes in T 2 values, the evolution of their relative amplitudes as a function of the sample water content and temperature was significantly different between samples (different homogeneous groups at 60 °C in Table 2) and helped to monitor the transformations within starch granules (Fig. 1a, b). Although the A(1) for the starch powder remained constant from 20 to 90 °C, that for hydrated SW samples tended to decrease during heating, with a similar slope for all the samples between 20 and 50 °C (Fig. 1a). A marked change of slope occurred between 50 and 60 °C from SW-40 to SW-50. The SW-35 sample evolved differently from the others, with no dramatic change in this temperature range. The linear decrease in A(1) in SW-35 demonstrated that probably most of the granules remained intact throughout the heating program for this sample, which is consistent with its insufficient hydration level for complete gelatinization. This continuous trend is in agreement with the evolution of A(2) for SW-35 which remained relatively stable even at high temperatures (Fig. 1b). The relative amplitude A(2) increased sharply for SW-40 to SW-45 between 50 and 60 °C, before decreasing at higher temperatures. Note that Fig. 1b displays a smaller ordinate scale (up to only 30%) to demonstrate its fluctuations better. SW-50 showed complex temperature-associated evolutions in A(2) amplitude. These changes were interpreted in terms of proton transfers from crystalline and constrained amylose structures (i.e., associated amylose chains and exterior chains of amylopectin, or amylose–lipid complexes), concomitantly with amylose leaching to the extra-granular water phase (Rondeau-Mouro et al. 2015). The different behavior observed for A(2) of SW-50 was probably a consequence of distinct water–starch interactions in this sample, which is consistent with the presence of a fifth T 2 component throughout the heating program, as discussed below.

T 2 relaxation time values (T 2(1) to T 2(4 + 5) in ms) and their relative amplitudes (A(1) to A(4 + 5) in %) (a–d) for wheat starch–water samples SW-35 (square), SW-40 (triangle), SW-42 (circle), SW-45 (diamond), and SW-50 (dash) samples as well as the relative amplitude A(1) of SP-11 sample (a) (dotted cube), as a function of temperature. The bars represent the standard deviation of the mean value. T 2 are presented in logarithmic scale
Consider now the evolution of the CPMG signal assigned to the water phase (Fig. 1c, d). Both the T 2(3) and T 2(4 + 5) relaxation times decreased between 20 and 50 °C and thus showed reduced mobility because of the water uptake and amylose leaching. This reduced mobility was most pronounced for the low hydrated sample, SW-35. Moreover, below 50 °C, the relative amplitude of intra-granular water A(3) was stable instead of decreasing (Curie’s law), reflecting that granules absorb water before gelatinization. Above 50 °C, the T 2(3) relaxation time increased, with a more marked increase starting from 50 °C for SW-50 than for the other samples (Fig. 1c). Less hydrated samples (SW-35 to SW-45) showed the increase in T 2(3) relaxation time only above 60 °C. Due to its long T 2(5) relaxation times measured even at low temperatures, the T 2(4 + 5) value of SW-50 was relatively high compared with the other samples (Fig. 1d). T 2(4 + 5) showed a sudden increase between 50 and 60 °C for SW-40 to SW-45, whereas these components decreased for SW-35 and SW-50. Further heating led to an increase in T 2(4 + 5), regardless of water content. Remarkably, in this temperature range, the mobility of extra-granular water T 2(4 + 5) for SW-50 was close to that of less hydrated samples (SW-45 to SW-40), which was due to the shortening of T 2(5) relaxation time with temperature in SW-50. The T 2(4 + 5) relaxation time was short compared with that of bulk water (∼2.5 s, under the same conditions), which can easily be explained by recalling that the dissolved starch molecules were very large in its native state, with high molecular weights of approximately 105–106 g/mol for amylose and above 107–109 g/mol for amylopectin. The solubilized starch material and leached amylose fragments being far lower in molecular weight (Greenwood 1979) are believed to greatly reduce water mobility in extra-granular space.
Across the 50–90 °C temperature range, it was particularly interesting to compare the evolutions of A(3) and A(4 + 5) because of their opposite trend, as shown in Fig. 1c, d. Between 50 and 60 °C, the intra-granular water phase characterized by A(3) diminished for SW-40 to SW-50 samples, probably due to a lower amount of available intra-granular water when granules disrupted. For temperatures higher than 60 °C, the intra-granular water mobility characterized by T 2(3) increased with temperature as expected, while its relative amplitude remained relatively stable for each sample, except for SW-35 which increased considerably between 60 and 90 °C. The curious behavior of the intra-granular water phase for SW-35 proved that less water prevented starch crystallite melting in this sample but, instead, enhanced further water absorption and granule swelling. This evolution is in accordance with the concomitant decrease in relative amplitude A(4 + 5) of the extra-granular water phase. The opposite trends between the A(3) and A(4 + 5) evolutions showed that starch and water transfer occurred between the water phases. A(4 + 5) represents a water phase containing starch constituents and fragments, which is supposed to become more and more viscous with the temperature increase (Donald 2004).
In order to compare the evolution of the relative amplitudes of T 2 components of each sample in a quantitative manner, the contribution of each T 2 to the total NMR signal was expressed as the mass intensity MI(i) (in volts per gram) as already explained in a previous study (Rondeau-Mouro et al. 2015). The mass intensities of each relaxation component of the SW starch–water samples were calculated and compared with those of starch powder MI(1) SP-11 (V/g of starch) and pure water MI(1) PW (V/g of water in sample). Wheat starch powder SP-11 was used as a reference for the comparison of decreases in MI(1 + 2) from the T 2(1 + 2) components between samples (Fig. 2a). The mass intensities of the short T 2(1) and T 2(1 + 2) components of SP and SW samples (MI(1) and MI(1 + 2), respectively) were calculated on the basis of the starch mass intensity in samples and interpreted as a function of temperature in terms of Curie’s law. Similarly, the mass intensity of the T 2(3 + 4 + 5) components in starch–water mixtures (MI(3 + 4 + 5)) was calculated on the basis of the mass intensity of pure water (PW) and presented as a function of the inverse of temperature (Fig. 2b). As shown in Fig. 2a, the mass intensity MI(1 + 2) for SP-11 and SW samples decreased linearly between 20 and 50 °C and obeyed Curie’s law, due to the magnetic susceptibility dependence on temperature. The magnitude of this decrease was expressed as the slope (in V·K/g) of the straight line joining the MI values as a function of temperature. The slope for MI(1 + 2) in SW samples (7855–8150 V·K/g) was close to that of SP-11 (7582 V·K/g), regardless of their water content. We noted that between 50 and 90 °C, the mass intensity decrease was no longer linear for SW samples and was highly dependent on the water content in the sample (Fig. 2a). This had already been demonstrated (Rondeau-Mouro et al. 2015) for a wheat starch–water mixture hydrated at 45% (wb), corresponding to the SW-45 sample in this study. The same procedure was applied for the longer T 2 components (3, 4, and 5), which were characteristics of the water phases in SW samples. Between 20 and 50 °C, SW samples displayed MI(3 + 4 + 5) slope values close to each other (16,016–17,006 V·K/g) and to that of pure water (15,479 V·K/g), regardless of the water content (Fig. 2b). Heating of starch–water mixtures above 40 °C led to an upward trend of MI(3 + 4 + 5) up to 80 °C, except for SW-35, for which MI(3 + 4 + 5) rose to only 60 °C and then decreased to 90 °C. The evolution of MI(3 + 4 + 5) curves differed from the MI(1) of pure water, which demonstrated that the signal measured for these protons did not obey Curie’s law, particularly above 50 °C.

Mass intensity (MI) of wheat starch–water SW-35 (square), SW-40 (triangle), SW-42 (circle), SW-45 (diamond), and SW-50 (dash) samples as a function of inverse of temperature of a solid components T 2(1 + 2) compared to the mass intensity of the solid component of wheat starch powder SP-11 (dotted cube) and b liquid components T 2(3 + 4 + 5) compared to the mass intensity of pure water PW (hatched cube)
In addition to this effect of temperature described by Curie’s law, the structure of starch granules dramatically changed during heating. To analyze and measure these changes quantitatively, the evolution of the contribution of the first component to the total NMR solid signal, noted C(1), was calculated as follows
Using Eq. 3, we estimated that at 20 °C, regardless of water content (35–50%), the first T 2 component represented on average 83.03% (±0.03) of the solid signal, as indicated in Fig. 3. This ratio might correspond to the expected amylopectin content of around 74.8%, recalculated taking into account the amount of protein as microconstituent (≤0.3% of protein) of the wheat starch powder used (BeMiller and Whistler 1996). The additional 8.2% not only might correspond to amylose chains and/or exterior chains of amylopectin that can form double helices but might also contain simple helices formed with endogenous lipids. Figure 3 shows that the water content had little influence on C(1) up to 50 °C, when it was 73.43% (±0.03) on average. If the crystalline amylopectin chain arrangements are considered to be retained even when amylose is leached out of the granules, this 10% decrease from the initial 83% was due to proton transfer to the second component A(2). This result confirms the assignment of T 2(1) to non-exchangeable CH protons from amylopectin and also to those from amylose which were transferred into the proton fraction T 2(2) during the swelling process (Rondeau-Mouro et al. 2015). Moreover, it indicates that amylose can form crystalline structures, as already evidenced in several NMR studies showing that in high amylose starches amylose forms double helices (Shi et al. 1998; Tester et al. 2000). Further heating above 50 °C led to different decreases in C(1) for samples hydrated above 35%. Indeed, C(1) calculated for SW-35 showed no great changes when the temperature rose.

Contribution C(1) to the total solid signal as a function of temperature in wheat starch–water SW-35 (square), SW-40 (triangle), SW-42 (circle), SW-45 (diamond), and SW-50 (dash) samples, C(1) expressed as percent with a mean standard deviation ≤5%, depending on water content and temperature
Knowing the intensity of the signal of the first and the second relaxation time components, I T2(1) and I T2(2), the mass intensity of the starch powder at each temperature measurement, and the total mass of each sample (m sample), the starch content could be calculated for each sample at each temperature as follows
Equation 4 was used to calculate the starch (or solid) content (expressed in %) at 20 °C and to follow its evolution at every temperature step, up to 90 °C. At 20 °C, SW-35, SW-40, SW-42, SW-45, and SW-50 samples were characterized by a solid content (SC) of 65.7% (±0.1), 59.6% (±0.7), 56.7% (±0.3), 55.7% (±0.1), and 47.8% (±1.0), respectively (Table 3). These values were close to the expected solid content in light of the water content in the samples (Table 1). This result demonstrates that NMR allowed determination of the water or starch content in each sample and followed their changes upon heating. The effects of the temperature that increase on the solid content of each SW hydrated sample could be evaluated in specific temperature ranges in relation to the endotherms measured in DSC (next section).
Finally and with the aim of correlating NMR and DSC measurements, T 2 relaxation times were also measured with a rapid heating rate (3 °C/min) for two samples, SW-45 and SW-50. As shown in Fig. 4 for SW-45 and Fig. 9 in Appendix for SW-50, the variation of the T 2 relaxation times and relative amplitudes determined by NMR indicated similar proton distribution and transfers between 20 and 90 °C regardless of the heating regime. The discrepancy between T 2(4 + 5) values in SW-50 came from the additional long T 2(5) component at low temperatures (Fig. 9 in Appendix).

Effects of heating regime on a A(1) to A(4 + 5) and b T 2(1) to T 2(4 + 5) as a function of temperature of SW-45. Dashed line denotes fast heating regime (3 °C/min), and solid line denotes slow heating regime (0.2 °C/min). T 2 are presented in logarithmic scale
Thermal Transitions Studied by DSC
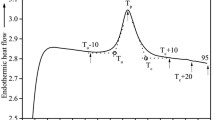
Figure 5 shows the DSC thermograms obtained for wheat starch–water mixtures (SW-35 to SW-50) and starch powder (SP-11), each heated from 20 to 130 °C at a rate of 3 °C/min. All SW samples featured three endotherms P1, P2, and P3, whereas a single endotherm emerged for the SP-11 sample. The results of DSC analyses are summarized in Table 4. To simplify interpretation, only four typical temperatures are reported (T o(P1), T p(P1), T p(P2), and T c(P2)). While T p(P1) and T p(P2) were the peak temperatures of the first and second transitions, T o(P1) and T c(P2) corresponded to the onset temperature of the P1 transition and the conclusion temperature of the P2 transition, respectively. The first two endothermic transitions, which have been named G (gelatinization) and M1 (the first melting transition) in the literature (Donovan and Mapes 1980), are commonly used when discussing starch gelatinization. Eliasson observed three peaks between 20 and 140 °C for wheat starch–water mixtures hydrated between 25.9 and 44% (wb). He showed that the temperature of the first and second endotherms were highly dependent on the water content (Eliasson 1980). Whereas the second transition peak could not be explained, the third transition was undoubtedly attributed to the melting of the amylose–lipid complex (Kugimiya et al. 1980). The third thermal transition, characterized by the P3 peak (Fig. 5), was not examined for all SW samples because of the insufficiently wide temperature range applied in the present experiments; however, this process was also dependent on water content in the sample, showing a trend similar to that of the P2 peak (increase in peak temperature with reducing water content).

DSC thermograms of wheat starch powder SP-11 and wheat starch–water samples, SW-35 to SW-50 heated at a scanning rate of 3 °C/min
In contrast to the onset temperature T o(P1), the conclusion temperature T c(P2) was more influenced by the water available in each sample. The temperature range of the first (P1) and the second peak (P2) transitions ∆T(P1 + P2) for hydrated samples was calculated and was found to vary between 52.6 °C (±1.0) and 41.8 °C (±2.0), going from the least hydrated (SW-35) to the most hydrated sample (SW-50). For hydrated samples, the first transition occurred in the temperature range 57.2 °C (±1.0)–70.3 °C (±0.3), with a peak temperature T p(P1) at 62.0 °C (±0.8) on average, whereas the second peak started at 72.4 °C (±1.0) and ended between 98.1 °C (±0.9) and 110.0 °C (±2.0), with an increase in peak temperature T p(P2) from 84.1 (±0.9) to 104.8 °C (±2.0) when the water content was reduced (50 to 35%). It is currently accepted that the phase transition of a single starch granule in excess of water might take place over a temperature range of 1–2 °C, whereas the gelatinization of the whole granule population occurs over a wider temperature range (Fredriksson et al. 1998). The gelatinization temperature may be defined as the point at which all, or a fraction of, the granules have undergone transformation (Lelievre 1976). This paradigm is probably due in part not only to the heterogeneity of starch granules but also to the diffuse melting process of complex macromolecular systems since starch is not a perfectly crystalline polymer, but rather a semi-crystalline structure (Lelievre 1974). Using a heating rate of 3 °C/min in our study, the temperature range of the first transition was at around 56–70 °C, depending on the sample water content, compared to that of Sasaki and Matsuki at around 50–66 °C, using a heating rate of 1 °C/min (Sasaki and Matsuki 1998). In our case, the temperature range was shifted to higher temperatures due to the faster heating rate used. This linear relationship between the transition temperature range and the heating rate was already demonstrated by Shiotsubo and Takahashi (Shiotsubo and Takahashi 1984). Thus, despite the differences attributed to the wheat starch source, the DSC results can also be affected by kinetics (heating rate) and different procedures, e.g., preparation of samples, time of equilibration, and size of sample. Similar to published results on starches of various botanical origins (Biliaderis et al. 1980; Garcia et al. 1996; Maache-Rezzoug et al. 2008), DSC thermograms recorded for wheat starch–water (SW) samples in the present study indicated that the peak temperature of the first transition (P1) was very slightly dependent on the water content, with a mean value of 62 °C. This temperature was close to that determined by Morales-Sanchez et al., who used a 5 °C/min heating rate and found the first peak of gelatinization at 61.2 °C (±0.1) for a wheat starch–water sample hydrated at 41.2% (wb) (Morales-Sanchez et al. 2009). In the present SW-42 sample, T p(P1) was 63.1 °C (±1.2). Further addition of water showed no significant differences in P1 peak temperature, as found for wheat starch–water samples hydrated at 50% (wb) showing a first transition at 59.0 °C (±0.4) by using a 10 °C/min heating rate (Tester and Sommerville 2001), which is close to the 61.2 °C (±1.3) for the present SW-50 sample.
The onset temperature of the first transition (P1) was 57.2 °C (±1.0) on average, while the conclusion temperature of the second transition (P2) was 110.2 °C (±1.0), 107.7 °C (±1.0), 104.5 °C (±4.0), 99.8 °C (±0.5), and 98.1 °C (±0.9) for SW-35, SW-40, SW-42, SW-45, and SW-50 samples, respectively (Table 4). These results are in agreement with those of Jankowski and Rha who reported 57.0 °C as T o and 108.0, 102.0, and 92.0 °C as T c for wheat starch–water samples containing 36.2, 43.3, and 48.8% of water (wb), respectively (Jankowski and Rha 1986). Obviously, the higher the water content, the lower the final temperature of disruption of crystalline regions. This is consistent with previous results showing that for limited water systems the temperature must be increased to achieve complete gelatinization (Eliasson and Larsson 1993).
The enthalpy change ∆H during the first and second transitions was highly dependent on the level of hydration, as shown in Fig. 6. The enthalpy changes for the overall gelatinization transition ∆H(P1 + P2) were found to be 5.8 (±0.7) J/g (dry basis (db)) for SW-50 and thus slightly higher than the value of 4.3 (±0.3) J/g (db) determined by Tester and Sommerville (2001). The area of gelatinization endotherms is indeed highly dependent on the amount of available water in wheat starch–water mixtures. Our results confirmed previous findings that the area of the P1 endotherm decreased when the water content was reduced, while the P2 endotherm moved to progressively higher temperatures, as shown in Table 4 and discussed in the next section (Eliasson 1980; Jankowski and Rha 1986; Tester and Sommerville 2001). Several models have been proposed in the literature to interpret the P1 and P2 thermal events; however, no model has been generally or universally accepted until now (Karapantsios et al. 2002). There are four main models describing the nature of the two-step endotherm in limited water conditions in which all the authors suggest that the enthalpy associated with gelatinization is the result of multiple thermal processes occurring within the same time frame (Donovan 1979; Evans and Haisman 1982; Slade and Levine 1988; Waigh et al. 2000). The influence of the amorphous starch regions on the enthalpy change has rarely been explained in any of these studies.

Enthalpy changes for the first (P1) and the second (P2) endotherms of wheat starch–water SW-35 to SW-50 samples as a function of their water content
According to Donovan, the first peak (P1) is considered to be induced by the swelling and dissolution of the amorphous regions, including a process of “stripping” of starch chains from the surface of crystallites, whereas the remaining crystallites are melted at higher temperatures which results in the P2 endotherm (Donovan 1979). Evans and Haisman argued that the successive endothermic peaks emerged due to melting transitions of crystalline materials with different stability, underlying the crystal imperfections and heterogeneous distribution of water within the sample (Evans and Haisman 1982). Granules with the least perfect crystallites gelatinize cooperatively, and consequently, the effective water concentration available for the remaining ungelatinized granules is reduced due to interactions between water and disordered polysaccharide chains. Slade and Levine reported that the most appropriate model for gelatinization includes a glass transition at P1 due to the plasticizing effect of water on the amorphous regions, and including the disordering of double helices associated with short-range order and a second phase transitions, P2, assigned to the disordering of amylopectin crystallites (Slade and Levine 1988). The fourth hypothesis was proposed by Waigh et al. and is close to the previous one, since the crystalline phase transitions (smectic to isotropic/nematic) are combined with plasticization of amorphous backbone regions (Waigh et al. 2000). They considered different levels of water content, a limiting water case for water content less than 28.6% (wb) corresponding to 40% on dry basis and a case of excess of water for water content greater than 28.6% (wb). In these conditions, they proposed that the P1 endotherm could be assigned to dissociation of double helices, while P2 may correspond to further helix–coil transitions, taking into account the possible reversibility of changes when samples were cooled down.
Correlations between NMR and DSC
A combined DSC and NMR approach has rarely been applied to wheat starch–water mixtures. Some of these studies investigated the effects of salts or sucrose on water mobility and starch gelatinization and showed complex patterns, depending on the constituent concentration (Chiotelli et al. 2002; Chinachoti et al. 1991). In other papers with joint observation of these two methods, the NMR results did not significantly improve the interpretation of thermal transitions observed by DSC. However, in a study on starches with different amylopectin content, Bosmans et al. showed a good correlation between the amplitude of the first NMR component and the DSC enthalpy of gelatinization (Bosmans et al. 2012). In the study reported here, the relaxation behavior of water and starch protons was studied at temperatures not higher than 90 °C to avoid water evaporation that would be critical from a quantitative point of view. By comparison, DSC experiments enabled us to study starch–water behavior over a wider temperature range, with a maximum temperature of 130 °C, thus allowing scanning of the gelatinization as a complete process. The DSC method also has the advantage of showing a continuous profile which can be used to determine the initial and the final temperature of transition, but with a lower time resolution and less quantitative information than NMR. It should be noted that the heating rate had no influence on the NMR results, as shown for SW-45 and SW-50 samples (Figs. 4 and 9 in Appendix, respectively). The T 2 relaxation times measured by NMR could thus be related to the DSC results. An example of the dependence of the DSC and NMR results on the heating regime is the study by Gonera and Cornillon, in which it was argued that the gelatinization range in the NMR measurements was narrower than in DSC (5 °C/min, linear change of temperature) due to stepwise change of temperature (0.1 °C/min) (Gonera and Cornillon 2002). However, they investigated highly hydrated samples (66.7% wb), and they started heating the samples only from 45 °C, so that they did not observe the initial swelling phase, which is an important step for gelatinization phenomena. We, therefore, consider that this statement is not applicable to our study, since the experimental conditions were not comparable.
Although the swelling of granules had already occurred at room temperature, more pronounced swelling took place with the heat uptake, coinciding with the onset temperature of gelatinization measured by DSC at about 56.2–57.6 °C, well below the peak temperature T p(P1) at about 62 °C. With regard to the NMR solid signal in FID, calculation of the loss of solid content (ΔSC) between 20 and 55 °C revealed that, on average, 12.3% (±0.7) of solid material was lost in this temperature range for SW-35 to SW-45, whereas a greater SC loss (16.3% (±2.0) on average) was found for the SW-50 sample (Table 3). As shown before, in the same temperature range, the contribution of the first component to the total NMR solid signal C(1) decreased by 10% for all samples. In the literature, crystallinity is exclusively associated with amylopectin (Ratnayake and Jackson 2008). However, the solid content is also dependent on the amylose content and this was clearly demonstrated by NMR measurements. Since the crystallinity is thought to be retained even when amylose is leached from the granule (D. French 1984), the decrease in C(1) up to 50 °C was assumed to originate from amylose transfer to the amorphous regions of granules concomitantly with the amylose leaching from the amorphous part and/or the crystallite surface into the extra-granular water phase (Rondeau-Mouro et al. 2015). This interpretation is in line with the hypothesis of Donovan who explained the first DSC endotherm by a process of stripping of the amorphous component initially present on the crystallite surface (Donovan 1979), which was also evidenced by Jenkins and Donald using a small angle neutron scattering (SANS) technique (Jenkins and Donald 1998). Moreover, Cooke and Gidley emphasized the importance of loss of molecular (double helical) order during heating observed concurrently with the loss of crystalline order (Cooke and Gidley 1992). In fact, this phenomenon could be related to the reversible swelling process of starch granules below 55 °C. All these previous studies support our results in which NMR helped to predict the extent of the release of amylose fragments (around 11.3–16.3% depending on the water content) before the beginning of the P1 transition. Table 3 demonstrates that for the most highly hydrated sample (SW-50), the amylose leaching was more effective than for the other samples. Since the volume outside the granules for SW-35 to SW-45 samples was smaller than that of the SW-50 sample, the latter showed a lower effect of close packing of starch granules, thus favoring the leaching of amylose fragments out of granules (Hermansson and Svegmark 1996). Moreover, the fact that this process was more significant for SW-50 can explain why even at 20 °C, we measured the long T 2(5) component. The latter can, therefore, be attributed to an additional water phase with low interaction with leached amylose. Although the onset temperature of P1 has often been discussed as the beginning of gelatinization, distinct from the melting process during P2, our NMR results demonstrated that polymer melting occurred during the first transition (P1), i.e., melting of amylopectin double helices and probably the helices made with amylose. Obviously, the more hydrated the sample, the greater the SC loss. Unlike the SW-35 sample, the SW-50 sample was characterized by the greater loss of solid content. Similarly, SW-50 displayed the highest ∆H(P1) value, whereas that of SW-35 starch was the lowest (Table 4). As shown in Fig. 7a, the enthalpy changes during the P1 transition presented the same trend as the loss of solid content between 55 and 70 °C, calculated using NMR results. The two straight lines ran almost parallel, thus suggesting that both amorphous and crystalline regions should be considered when discussing the occurrence of the first transition peak. In the 55–70 °C temperature range, melting of starch crystallites occurred as a result of heat uptake in the presence of sufficient water, and for the same reasons, the amylose leaching was enhanced. By extrapolation, the straight lines of the enthalpy ∆H(P1) and SC loss to zero meant that the minimum water content required to initiate gelatinization could be estimated. According to Fig. 7a, this value was 24.5% from DSC results and 23.7% from the NMR results, which is in good agreement with the 25% water content (wb) proposed by Lund and Wirakartakusumah (1984). The same result was suggested by Eliasson (1980) and Wootton and Bumunuarachchi (1979), who found 24.8% (33%, db) and 24.2% (32%, db) as the minimum water content (wb), respectively. Since the water should be in sufficient quantity to provide adequate hydration for gelatinization (Biliaderis 1992), it was proposed that at water concentrations lower than 61% (wb), the completion of hydration-assisted dissociation of double helices would not be possible. Moreover, Spigno and De Faveri showed that activation energy for gelatinization decreased with the increase in water content and stabilized at about 60% (wb) of water content (Spigno and De Faveri 2004). Considering the water content range in our study (35–50% wb), it is clear that starch granules in all samples were only partially gelatinized, which was confirmed by further but still partial loss of solid content in samples between 55 and 70 °C, calculated using NMR results (Table 3). Figure 7b demonstrates the good correlation between the enthalpy changes of P1 endotherm (∆H(P1)) and the loss of solid content (∆SC) for each sample during the first transition P1 (55–70 °C). Highly significant Pearson’s correlation coefficients were calculated at +0.9833 with a probability of 0.0054.

a Enthalpy changes of P1 endotherm ∆H(P1) and loss of solid content ∆SC during the first transition P1 (55–70 °C) and b linear correlation between ∆H(P1) and ∆SC of wheat starch–water SW-35 to SW-50 samples as a function of their water content
The NMR results in the present study thus agreed with the interpretations of DSC endotherms postulated by Waigh et al. in the case of limited water content, wherein two DSC endotherms were measured because of the two-step process, i.e., smectic to isotropic transitions at lower temperature followed at higher temperatures by the helix–coil transitions. Moreover, going from SW-50 to SW-35, the ∆H(P1) transition decreased, because of the reduction of water, in contrast to P2 transition (Fig. 6). This opposite trend of the P1 and P2 enthalpy transitions has already been reported by some authors (Donovan 1979; Tananuwong and Reid 2004) and should be taken into account when discussing the occurrence of bimodal transitions during gelatinization. It can be supposed that this thermal bimodality is related to bimodal distribution of granules, as the A- and B-type granules differed both in composition and in properties (Soulaka and Morrison 1985). Small granules of wheat starch tend to be more crystalline than large granules; unlike the latter, the former have greater swelling capacities probably because of their lower amount of lipids (Wong and Lelievre 1982). Additionally, the starch granules of cereals (such as maize, wheat, and rice) are characterized by compact A-type crystallites and thus should have better thermal stability (Ambigaipalan et al. 2014; Donovan et al. 1983), as opposed to B-type crystallites, characteristic of tubers, fruits, and stem starches (such as potato, banana, and sago starches). Indeed, A- and B-type starches have shorter and longer amylopectin helices, respectively, and changes in the lamellar structure, which consists of alternating regions of crystalline and amorphous material, should correspond to a two-step process (Waigh et al. 2000). With an increase in temperature, the mobility of the amylopectin double helices is increased and a smectic to isotropic phase (A-type starches) or smectic to nematic (B-type starches) transition occurs. Further heating leads to the helix–coil transition by the unwinding of helices. As proposed by Waigh et al., the two DSC endotherms could thus be ascribed to these two transitions (Waigh et al. 2000). Since the first transition P1 was highly correlated with the crystallite melting measured by NMR, Donovan’s model proposing that crystallites melt at higher temperatures into the P2 transition seemed not to be applicable to explain our results (Donovan 1979). On the other hand, the two-step models of gelatinization proposed by Evans and Haisman, who considered successive endothermic peaks as melting transitions of crystalline materials showing various imperfections and heterogeneous distribution of water, do fit with our results (Evans and Haisman 1982). However, the TD-NMR method cannot help to distinguish between perfect and imperfect crystallites in order to analyze this hypothesis but, instead, allow monitoring of the water distribution and starch transformations under heating.
The third interpretation which takes into account the glass transition of starch and two phase transitions within amylopectin crystallites (Slade and Levine 1988) was difficult to analyze with our NMR experimental setup, i.e., temperature range from 20 to 90 °C, and starch–water mixture using low to intermediate hydration levels (Fig. 8). In our experimental conditions, only the starch powder (SP-11) could not gelatinize due to the insufficient water level (11% wb) but, instead, showed a single endothermic transition at 61.9 °C (±0.3) (Fig. 5), which was characterized by a ∆H of 0.9 (±0.2) J/g and appeared probably due to enthalpy relaxation phenomena, which had already been observed in the range 50–64 °C for low hydrated starch powder from corn (Shogren 1992) or for starch lintners from potato (Le Bail et al. 1993).
If we take into account the second P2 transition and previous interpretations, the crystallinity in gelatinized granules should be totally lost close to the conclusion temperature T c(P2). However, in the present study, no comparison of the P2 transition with NMR results was possible, as the temperature range between 70 and 90 °C did not cover the whole P2 phase transition, especially for samples with a low hydration level.
Conclusion
In this study, we investigated how the water content (11–50% wb) in starch affected the water distribution and interactions with starch during its swelling and gelatinization. NMR measurements showed that, despite temperature changes, the water content is of major importance for the understanding of the proton transfer mechanisms between each constituent. The T 2 results between 20 and 55 °C revealed that the SW-50 sample leached out more amylose fragments than less hydrated samples. The more pronounced leaching can explain the longest T 2 component, T 2(5), already found at 20 °C, which was attributed to an additional water phase with low interaction with leached amylose.
This study also addressed the value of combining multiple analytical techniques, whereby NMR was used in combination with DSC to improve understanding of starch gelatinization. The DSC analysis confirmed that the gelatinization mechanisms depend on water availability. To date, enthalpy changes have been used as an indicator of the amount of crystallinity of the sample being analyzed (enthalpy of fusion). The combined approach used in the present study indicated that the degree to which the protons of starch–water mixtures were transferred from the solid to the liquid phase fitted well with the enthalpy changes that the sample had undergone during the first phase transition. This relationship helped to explain the existence of the first gelatinization endotherm, as we showed that the solid loss from both crystalline and amorphous regions should be considered. At the same time, we emphasized the importance of the reversible swelling of starch granules and the leaching of amylose fragments, thus supporting the concept that the amorphous phase in starch granules has an important role in gelatinization mechanisms.
References
Ambigaipalan, P., Hoover, R., Donner, E., & Liu, Q. (2014). Starch chain interactions within the amorphous and crystalline domains of pulse starches during heat-moisture treatment at different temperatures and their impact on physicochemical properties. Food Chemistry, 143, 175–184.
Assifaoui, A., Champion, D., Chiotelli, E., & Verel, A. (2006). Rheological behaviour of biscuit dough in relation to water mobility. International Journal of Food Science & Technology, 41(s2), 124–128.
Atwell, W. A. (2001). Composition of commercial flour. Wheat Flour, 27–45.
BeMiller, J. N., & Whistler, R. L. (1996). Carbohydrates. In O. R. Fennema et al. (Eds.), Food chemistry (3rd ed., pp. 191–204). New York: Taylor and Francis Group.
Biliaderis, C. (1992). Structures and phase transitions of starch in food systems. Food Technology, 46(6), 98–109.
Biliaderis, C., Maurice, T., & Vose, J. (1980). Starch gelatinization phenomena studied by differential scanning calorimetry. Journal of Food Science, 45(6), 1669–1674.
Bogracheva, T. Y., Wang, Y., & Hedley, C. (2001). The effect of water content on the ordered/disordered structures in starches. Biopolymers, 58(3), 247–259.
Bosmans, G. M., Lagrain, B., Deleu, L. J., Fierens, E., Hills, B. P., & Delcour, J. A. (2012). Assignments of proton populations in dough and bread using NMR relaxometry of starch, gluten, and flour model systems. Journal of Agricultural and Food Chemistry, 60(21), 5461–5470.
Bosmans, G. M., Pareyt, B., & Delcour, J. A. (2016). Non-additive response of blends of rice and potato starch during heating at intermediate water contents: a differential scanning calorimetry and proton nuclear magnetic resonance study. Food Chemistry, 192, 586–595.
Chinachoti, P., Kimshin, M. S., Mari, F., & Lo, L. (1991). Gelatinization of wheat-starch in the presence of sucrose and sodium-chloride - correlation between gelatinization temperature and water mobility as determined by oxygen-17 nuclear-magnetic-resonance. Cereal Chemistry, 68(3), 245–248.
Chiotelli, E., Pilosio, G., & Le Meste, M. (2002). Effect of sodium chloride on the gelatinization of starch: a multimeasurement study. Biopolymers, 63(1), 41–58.
Choi, S.-G., & Kerr, W. L. (2003). 1H NMR studies of molecular mobility in wheat starch. Food Research International, 36(4), 341–348.
Considine, D. M., & Considine, G. D. (1982). Bread and bakery products. In Foods and food production encyclopedia (pp. 282-295). New York: Van Nostrand Reinhold Co.
Cooke, D., & Gidley, M. J. (1992). Loss of crystalline and molecular order during starch gelatinisation: origin of the enthalpic transition. Carbohydrate Research, 227, 103–112.
Da Silva, C. M., Ciacco, C., Barberis, G., Solano, W., & Rettori, C. (1996). Starch gelatinization measured by pulsed nuclear magnetic resonance. Cereal Chemistry, 73(3), 297–301.
Donald, A. (2004). Understanding starch structure and functionality. In A.-C. Eliasson (Ed.),Starch in food: structure, function and applications (pp. 156–184). Boca Raton: Woodhead Publishing Limited.
Donovan, J. (1979). Phase transitions of the starch–water system. Biopolymers, 18(2), 263–275.
Donovan, J., & Mapes, C. (1980). Multiple phase transitions of starches and Nägeli amylodextrins. Starch-Stärke, 32(6), 190–193.
Donovan, J., Lorenz, K., & Kulp, K. (1983). Differential scanning calorimetry of heat-moisture treated wheat and potato starches. Cereal Chemistry, 60(5), 381-387.
Eliasson, A.-C. (1980). Effect of water content on the gelatinization of wheat starch. Starch-Stärke, 32(8), 270–272.
Eliasson, A.-C., & Larsson, K. (1993). Cereals in breadmaking: a molecular colloidal approach. New York: Marcel Dekker.
Evans, I., & Haisman, D. (1982). The effect of solutes on the gelatinization temperature range of potato starch. Starch-Stärke, 34(7), 224–231.
Fredriksson, H., Silverio, J., Andersson, R., Eliasson, A.-C., & Åman, P. (1998). The influence of amylose and amylopectin characteristics on gelatinization and retrogradation properties of different starches. Carbohydrate Polymers, 35(3), 119–134.
French, D. (1972). Fine structure of starch and its relationship to the organization of starch granules. 澱粉科学, 19(1), 8–25.
French, D. (1984). Organization of starch granules. Starch: chemistry and technology, 2, 183–247.
Garcia, V., Colonna, P., Lourdin, D., Buleon, A., Bizot, H., & Ollivon, M. (1996). Thermal transitions of cassava starch at intermediate water contents. Journal of Thermal Analysis and Calorimetry, 47(5), 1213–1228.
Godet, M., Bizot, H., & Buléon, A. (1995). Crystallization of amylose—fatty acid complexes prepared with different amylose chain lengths. Carbohydrate Polymers, 27(1), 47–52.
Gonera, A., & Cornillon, P. (2002). Gelatinization of starch/gum/sugar systems studied by using DSC, NMR, and CSLM. Starch-Stärke, 54(11), 508–516.
Greenwood, C. (1979). Observations on the structure of the starch granule. In J. M. V. Blanshard and J. R. Mitchell (Ed.), Polysaccharides in Food (pp. 129-138). London: Butterworths.
Hermansson, A.-M., & Svegmark, K. (1996). Developments in the understanding of starch functionality. Trends in Food Science & Technology, 7(11), 345–353.
Hills, B., Godward, J., Manning, C., Biechlin, J., & Wright, K. (1998). Microstructural characterization of starch systems by NMR relaxation and Q-space microscopy. Magnetic Resonance Imaging, 16(5), 557–564.
Hizukuri, S. (1986). Polymodal distribution of the chain lengths of amylopectins, and its significance. Carbohydrate Research, 147(2), 342–347.
Hoseney, R. C. (1994). Soft wheat products. In Principles of cereal science and technology, (pp. 275-306). St. Paul: American Association of Cereal Chemists.
Jankowski, T., & Rha, C. (1986). Differential scanning calorimetry study of the wheat grain cooking process. Starch-Stärke, 38(2), 45–48.
Jenkins, P. J., & Donald, A. M. (1998). Gelatinisation of starch: a combined SAXS/WAXS/DSC and SANS study. Carbohydrate Research, 308(1), 133–147.
Karapantsios, T., Sakonidou, E., & Raphaelides, S. (2002). Water dispersion kinetics during starch gelatinization. Carbohydrate Polymers, 49(4), 479–490.
Kugimiya, M., Donovan, J., & Wong, R. (1980). Phase transitions of amylose-lipid complexes in starches: a calorimetric study. Starch-Stärke, 32(8), 265–270.
Le Bail, P., Bizot, H., & Buléon, A. (1993). ‘B’to ‘A’type phase transition in short amylose chains. Carbohydrate Polymers, 21(2), 99–104.
Le Botlan, D., Rugraff, Y., Martin, C., & Colonna, P. (1998). Quantitative determination of bound water in wheat starch by time domain NMR spectroscopy. Carbohydrate Research, 308(1), 29–36.
Le Grand, F., Cambert, M., & Mariette, F. (2007). NMR signal analysis to characterize solid, aqueous, and lipid phases in baked cakes. Journal of Agricultural and Food Chemistry, 55(26), 10947–10952.
Lelievre, J. (1974). Starch gelatinization. Journal of Applied Polymer Science, 18(1), 293–296.
Lelievre, J. (1976). Theory of gelatinization in a starch-water-solute system. Polymer, 17(10), 854–858.
Lund, D., & Wirakartakusumah, M. (1984). A model for starch gelatinization phenomena. Engineering and Food, 1, 425–432.
Maache-Rezzoug, Z., Zarguili, I., Loisel, C., Queveau, D., & Buleon, A. (2008). Structural modifications and thermal transitions of standard maize starch after DIC hydrothermal treatment. Carbohydrate Polymers, 74(4), 802–812.
MacArthur, L., & D’appolonia, B. (1979). Comparison of oat and wheat carbohydrates. II. Starch. Cereal Chemistry, 56, 458–461.
Mariette, F., Guillement, J.P., Tellier, C., & Marchal, P. (1996). Continuous relaxation time distribution decomposition by MEM, Chapter 10.
Marquardt, D. W. (1963). An algorithm for least-squares estimation of nonlinear parameters. Journal of the Society for Industrial & Applied Mathematics, 11(2), 431–441.
Medcalf, D., & Gilles, K. (1965). Wheat starches. I. Comparison of physicochemical properties. Cereal Chemistry, 42(558), I965.
Meiboom, S., & Gill, D. (1958). Modified spin-echo method for measuring nuclear relaxation times. Review of Scientific Instruments, 29(8), 688–691.
Morales-Sanchez, E., Figueroa, J., & Gaytan-Martínez, M. (2009). Wet method for measuring starch gelatinization temperature using electrical conductivity. Journal of Food Science, 74(7), E382–E385.
Pojić, M., Musse, M., Rondeau, C., Hadnađev, M., Grenier, D., Mariette, F., et al. (2016). Overall and local bread expansion, mechanical properties, and molecular structure during bread baking: effect of emulsifying starches. Food and Bioprocess Technology, 1–19.
Ratnayake, W. S., & Jackson, D. S. (2008). Starch gelatinization. Advances in Food and Nutrition Research, 55, 221–268.
Rondeau-Mouro, C., Cambert, M., Kovrlija, R., Musse, M., Lucas, T., & Mariette, F. (2015). Temperature-associated proton dynamics in wheat starch-based model systems and wheat flour dough evaluated by NMR. Food and Bioprocess Technology, 8, 777–790.
Sasaki, T., & Matsuki, J. (1998). Effect of wheat starch structure on swelling power. Cereal Chemistry, 75(4), 525–529.
Schirmer, M., Jekle, M., & Becker, T. (2015). Starch gelatinization and its complexity for analysis. Starch-Stärke, 67(1–2), 30–41.
Shi, Y.-C., Capitani, T., Trzasko, P., & Jeffcoat, R. (1998). Molecular structure of a low-amylopectin starch and other high-amylose maize starches. Journal of Cereal Science, 27(3), 289–299.
Shiotsubo, T., & Takahashi, K. (1984). Differential thermal analysis of potato starch gelatinization. Agricultural and Biological Chemistry, 48(1), 9–17.
Shogren, R. (1992). Effect of moisture content on the melting and subsequent physical aging of cornstarch. Carbohydrate Polymers, 19(2), 83–90.
Singh, N., Singh, J., Kaur, L., Singh Sodhi, N., & Singh Gill, B. (2003). Morphological, thermal and rheological properties of starches from different botanical sources. Food Chemistry, 81(2), 219–231.
Slade, L., & Levine, H. (1988). Non-equilibrium melting of native granular starch: part I. Temperature location of the glass transition associated with gelatinization of A-type cereal starches. Carbohydrate Polymers, 8(3), 183–208.
Soulaka, A. B., & Morrison, W. R. (1985). The amylose and lipid contents, dimensions, and gelatinisation characteristics of some wheat starches and their A-and B-granule fractions. Journal of the Science of Food and Agriculture, 36(8), 709–718.
Spigno, G., & De Faveri, D. M. (2004). Gelatinization kinetics of rice starch studied by non-isothermal calorimetric technique: influence of extraction method, water concentration and heating rate. Journal of Food Engineering, 62(4), 337–344.
Svensson, E., & Eliasson, A.-C. (1995). Crystalline changes in native wheat and potato starches at intermediate water levels during gelatinization. Carbohydrate Polymers, 26(3), 171–176.
Tananuwong, K., & Reid, D. S. (2004). DSC and NMR relaxation studies of starch–water interactions during gelatinization. Carbohydrate Polymers, 58(3), 345–358.
Tang, H.-R., Godward, J., & Hills, B. (2000). The distribution of water in native starch granules—a multinuclear NMR study. Carbohydrate Polymers, 43(4), 375–387.
Tang, H.-R., Brun, A., & Hills, B. (2001). A proton NMR relaxation study of the gelatinisation and acid hydrolysis of native potato starch. Carbohydrate Polymers, 46(1), 7–18.
Tester, R., & Sommerville, M. (2001). Swelling and enzymatic hydrolysis of starch in low water systems. Journal of Cereal Science, 33(2), 193–203.
Tester, R., Debon, S., & Sommerville, M. (2000). Annealing of maize starch. Carbohydrate Polymers, 42(3), 287–299.
Waigh, T. A., Gidley, M. J., Komanshek, B. U., & Donald, A. M. (2000). The phase transformations in starch during gelatinisation: a liquid crystalline approach. Carbohydrate Research, 328(2), 165–176.
Wild, D., & Blanshard, J. (1986). The relationship of the crystal structure of amylose polymorphs to the structure of the starch granule. Carbohydrate Polymers, 6(2), 121–143.
Wong, R., & Lelievre, J. (1982). Comparison of the crystallinities of wheat starches with different swelling capacities. Starch-Stärke, 34(5), 159–161.
Wootton, M., & Bamunuarachchi, A. (1979). Application of differential scanning calorimetry to starch gelatinization. I. Commercial native and modified starches. Starch-Stärke, 31(6), 201–204.
Xie, F., Halley, P. J., & Avérous, L. (2012). Rheology to understand and optimize processability, structures and properties of starch polymeric materials. Progress in Polymer Science, 37(4), 595–623.
Acknowledgements
This work was performed using the NMR facilities of the PRISM Research Platform (Rennes, France). The authors thank the Regional Council of Brittany for their financial support. We would also like to thank Denis Lourdin and Marion De Carvalho (INRA Nantes, France) for carrying out the DSC analysis of the starch samples.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix

T 2 (ms) distributions at 20 °C (black diamond) and 60 °C (white diamond) of SW-45 (logarithmic scale)

Effects of heating regime on a A(1) to A(4 + 5) and b T 2(1) to T 2(4 + 5) as a function of temperature of wheat starch–water SW-50 sample (logarithmic scale). Dashed line denotes fast heating regime (3 °C/min), and solid line denotes slow heating regime (0.2 °C/min)
Rights and permissions
About this article

Cite this article
Kovrlija, R., Rondeau-Mouro, C. Hydrothermal Changes of Starch Monitored by Combined NMR and DSC Methods. Food Bioprocess Technol 10, 445–461 (2017). https://doi.org/10.1007/s11947-016-1832-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11947-016-1832-9