
Abstract
Purpose of Review
The purpose of this review is to summarize currently available and developing diagnostic and treatment options for hereditary transthyretin amyloid polyneuropathy. Transthyretin amyloidosis (ATTR) predominantly manifests with cardiomyopathy and/or peripheral neuropathy, but amyloid deposits may be found in other organs or tissues.
Recent Findings
Currently available treatments include transthyretin gene silencers (for hereditary ATTR peripheral neuropathy only) and transthyretin stabilizers (tafamidis for ATTR cardiomyopathy in the USA, and for both hereditary ATTR peripheral neuropathy and ATTR cardiomyopathy in Europe, Japan, Brazil, and some other countries), and liver transplantation. Gene silencers stop the progression of hereditary ATTR peripheral neuropathy in most patients, and transthyretin stabilizers reduce hospitalizations and mortality in patients with ATTR cardiomyopathy. The use of liver transplantation for ATTR has declined with the availability of more effective therapies, and shortage of available allografts. On the horizon are new treatments already in clinical trials including new gene silencers and gene editing agents, new transthyretin stabilizers, and amyloid removal treatments.
Summary
Recently approved treatments for ATTR have changed its natural history, and additional medications may get approved in the near future. Early diagnosis is still essential to improve treatment outcomes. New management strategies may include combinations of gene silencers, transthyretin stabilizers, gene editing, and amyloid removal agents, but the cost may become the limiting factor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic amyloidoses are a group of rare conditions associated with tissue deposition of insoluble deposits of misfolded protein leading to tissue injury and dysfunction [1]. Two most common types are light-chain amyloidosis (AL) and transthyretin amyloidosis (ATTR). ATTR may present as a hereditary or variant ATTR (hATTR or ATTRv) or wild-type ATTR (wtATTR) with normal transthyretin (TTR) genotype [2]. hATTR is a hereditary autosomal dominant disease with pathogenic mutations of TTR gene manifesting primarily with cardiomyopathy or peripheral neuropathy, and with rare mutations leading to oculoleptomeningeal ATTR (OLMA). In contrast, wtATTR manifests primarily as cardiomyopathy [3], although wtATTR deposits can be found in connective and musculoskeletal tissue as well [4]. Patients with wtATTR may also develop peripheral neuropathy symptoms, but it is usually not a major manifestation of wtATTR [5,6,7].
Diagnosis
Early and accurate diagnosis of hATTR-associated peripheral neuropathy (hATTR-PN, previously known as familial amyloid polyneuropathy type I or FAP) is essential for timely treatment and effective management as currently available treatments slow down or stop the progression of disease with few patients achieving mostly mild clinical improvement [8, 9••, 10••]. Unfortunately, the diagnosis of hATTR is often substantially delayed by 1.2 to 2.9 years after the onset of symptoms [11, 12]. Typical clinical phenotype of both AL or hATTR-associated polyneuropathy consists of progressive painful sensorimotor polyneuropathy with dysautonomia [13]. Diagnosis is based on clinical presentation, genetic testing, histopathologic evidence of amyloidosis, and amyloid typing. Dysautonomia may be initially subtle or absent, especially with late-onset hATTR-PN (after age of 50) [14]. In the USA and in most parts of the world outside of endemic areas, late-onset hATTR-PN is much more prevalent than early-onset hATTR-PN.
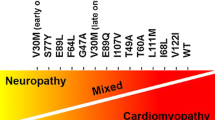
Multisystemic involvement and associated clinical symptoms reflect amyloid tissue deposition in various organs and tissues, and the recognition of “red flag” symptoms may facilitate earlier diagnosis [15, 16]. Initial attempts to use artificial intelligence to identify hATTR patients in electronic medical records were driven by retrieval of “red flag” symptoms and identified additional 48% of patients who should undergo genetic testing for possible hATTR [17]. Clinical progression of hATTR-PN is often rapid and patients get disabled within 5–7 years [18••]. There is no pathognomonic electrodiagnostic pattern for hATTR-PN and the most frequent phenotype is axonal sensorimotor polyneuropathy, but rarely demyelinating features might prevail increasing the risk of possible misdiagnosis as autoimmune or hereditary demyelinating neuropathies [19]. Carpal tunnel syndrome often precedes other clinical manifestations of amyloid deposition and histopathologic examination of tenosynovium after carpal tunnel surgery may reveal different types of amyloid deposits (AL, hATTR, wtATTR) with some patients already having unrecognized amyloid cardiomyopathy [20]. There is an increased risk of cardiac amyloidosis in hATTR patients after CTS, typically occurring between 5 and 9 years later [21]. Similarly, CTS often precedes the onset of hATTR-PN as well [22]. Routine use of nerve biopsy for diagnosing hATTR-PN is limited by its invasiveness, procedure-related morbidity, and limited diagnostic sensitivity. Neuroimaging of amyloid neuropathy with magnetic resonance neurography and high-resolution neuromuscular ultrasound may demonstrate nerve enlargement (thickening) that is not specific for amyloidosis, but may be helpful to detect early abnormalities in previously asymptomatic TTR mutation carriers [23, 24]. In amyloid cardiomyopathy, cardiac scintigraphy is relatively specific for diagnosing cardiac deposition of ATTR non-invasively, and it may demonstrate ATTR in skeletal muscle as well [25, 26]. PET/MRI using amyloid ligands may also reveal nerve uptake and facilitate nerve biopsy [27]. In the central nervous system (CNS), PET may demonstrate amyloid deposition in patients with OLMA [28••]. As Congo red staining on the tissue is not specific for the type of amyloid, typing of amyloid is required to determine the underlying disorder [29]. Amyloid deposits may be demonstrated and typed in different types of non-neural tissue including accessory salivary glands, abdominal or perirectal fat, bone marrow, and gastrointestinal tract [30]. Mass spectrometric-based proteomic analysis is considered a gold standard in amyloid typing and is considered superior to immunohistochemistry, but its availability may be limited in some geographic locations due to the cost and the lack of access [31]. Skin biopsy is increasingly used in evaluation of small fiber neuropathy and may be helpful in documenting new onset of nerve injury in previously identified carriers of TTR mutations, while cutaneous amyloid may be detected in up to 86% of patients with hATTR polyneuropathy [32, 33]. Loss of epidermal nerve fibers on skin biopsies was reported in asymptomatic (or presymptomatic) patients with early-onset p.V50M ATTR contrasting normal biopsies of asymptomatic patients with late-onset p.V50M ATTR [33]. Mutation p.V50M is also known as V30M when 20 amino acid sequence of signal peptide is omitted from TTR. Phenotype of ATTR manifestations is to some degree determined by TTR genotype, with substantial phenotypic variability, even within the same family. The role of genetic testing in patients with progressive neuropathy and suspected hATTR is well established, but genetic counseling should precede potential screening of asymptomatic suspected mutation carriers [34]. Delayed onset of hATTR in many patients and its limited penetrance warrant clinical follow-up of mutation carriers. Asymptomatic patients should not be treated preventively with currently available treatments but early treatment following symptom onset is essential to improve outcomes [35•]. However, at this time, there is still limited data on the optimal follow-up regimen of carriers and on appropriate timing for starting early treatment beyond clinically manifesting amyloid neuropathy and/or cardiomyopathy [34, 36].
Treatment of hATTR-PN
Current treatment of hATTR-PN relies on two primary strategies, TTR reduction and tetramer stabilization. Reduction of circulating TTR limits the building blocks for amyloid production while stabilization of TTR tetramer reduces dissociation into monomeric TTR, the form of TTR that is prone to misfolding and amyloid aggregation. Most of TTR is produced in the liver, with some production by the retinal pigment epithelium and choroid plexus in the CNS. Until recent years, the main strategy to limit TTR production was liver transplantation [37]. In 2011, TTR stabilizer tafamidis was approved for treatment of early stages of hATTR-PN in Europe, but it did not get approved in the USA (Table 1). Development of “gene silencers” selectively targeting the production of TTR in hepatocytes allowed effective suppression of circulating levels of both variant and wild-type TTR [38]. In 2018, the first two gene silencers that effectively suppress TTR production were approved by the FDA for treatment of hATTR-PN, patisiran and inotersen, followed by another gene silencer vutrisiran in 2022 (Table 1). In pivotal phase III clinical trials, all gene silencers resulted in significant reduction of circulating TTR levels. Clinical studies with TTR gene silencers enrolled ambulatory patients with polyneuropathy disability (PND) stages 1/2/3 and FAP 1/2 (Table 2), as advanced stage non-ambulatory patients with PND stage 4 or FAP 3 were not expected to have significant clinical benefits.
Gene silencers
Patisiran
Patisiran is a double-stranded TTR directed small interfering RNA (siRNA) encapsulated in a lipid nanoparticle. In hepatocytes, the siRNA is incorporated into the inactive RNA-induced silencing complex (RISC). Within the RISC, the antisense strand of siRNA binds to the 3′-untranslated region of TTR messenger RNA (mRNA). This binding region is genetically conserved and present in both wild-type and variant disease, regardless of the specific disease-causing mutation. Transthyretin mRNA is then cleaved, resulting in lowered TTR production. In the pivotal APOLLO study (NCT01960348), hATTR-PN patients were randomized 2:1 to 0.3 mg/kg patisiran versus placebo IV every 3 weeks for 18 months. The primary outcome was the mNIS + 7, a validated scale of hATTR-PN severity with higher scores indicating greater disability [39]. The least mean square change in mNIS + 7 among treated patients was − 6.0 ± 1.7 versus 28.0 ± 2.6 in placebo treated patients [9••]. Over 18 months, the median TTR reduction in treated patients was 81%. Significant differences were also noted in secondary quality of life outcomes at 18 months as measured by the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QOL-DN) questionnaire. Adverse events that were more common in patisiran patients included peripheral edema and infusion-related reactions, and treatment-related discontinuations were more common in the placebo group [9••]. As with other gene silencers, patients are recommended to take vitamin A supplementation as gene silencing of TTR reduces vitamin A levels as well.
Inotersen
Inotersen, is a 2′-O-methoxyethyl-modified antisense oligonucleotide (ASO) RNA molecule that binds to TTR mRNA and results in its degradation via Ribonuclease H (RNAseH). In turn, this degradation lowers TTR production. In the NEURO-TTR study, inotersen was tested with patients randomized 2:1 to inotersen 300 mg subcutaneously weekly or placebo over 66 weeks (NCT01737398). Results were qualitatively similar to patisiran. For mNIS + 7, the least squares mean difference between the treatment and placebo groups was − 19.7 points (95% confidence interval, − 26.4 to − 13.0) favoring treatment at 66 weeks [10••]. The Norfolk QOL-DN scores also favored treatment. Inotersen reduced circulating TTR levels as expected with median TTR nadir reduction of 79.0% compared to baseline. Adverse events that were more common in the inotersen group included nausea, pyrexia, chills, vomiting, anemia, thrombocytopenia, and lowered platelet counts [10••]. Due to the risk of rare but serious adverse events of hemorrhage and glomerulonephritis seen in clinical testing, in the USA, inotersen must be prescribed under a risk evaluation and mitigation strategy (REMS) program [40].
Vutrisiran
In 2022, vutrisiran, an siRNA conjugated to a triantennary N-acetylgalactosamine (GalNAc) ligand, was approved. Vutrisiran binds the asialoglycoprotein receptor on hepatocytes and similarly to patisiran reduces circulating TTR through RNA interference. The GalNAc conjugate enhances stability and reduces the need for dosing to every 3 months via subcutaneous injection. In the phase III HELIOS-A study with patisiran as an active comparator arm and an external placebo comparison group from APOLLO, vutrisiran patients experienced no significant worsening in mNIS + 7 at 9 months compared to baseline, with a − 17.00 point (95% CI, − 21.78 to − 12.22) difference in mNIS + 7 compared to APOLLO placebo patients (NCT03759379). Most adverse events were mild or moderate in severity and were not clearly increased compared to placebo [41••].
TTR stabilizers
Tafamidis
In the USA, no TTR stabilizers are approved by the FDA for treatment of hATTR-PN, though tafamidis, a small molecule benzoxazole derivative, has been approved since 2011 for use in mild hATTR-PN in Europe and many other countries dosed at 20 mg a day orally (Table 1). FDA approval in the USA was not granted for tafamidis in hATTR-PN due to insufficient evidence of clinical efficacy [8], but the drug was approved for ATTR-CM in the USA in 2019. Combination treatment with stabilizers and siRNA or inotersen has not been rigorously studied but is employed by some physicians in clinical practice, especially in patients with both hATTR-PN and hATTR-CM.
Liver transplantation
By removing the native liver and replacing with a transplanted liver, the patient’s primary source of variant TTR is removed and further systemic TTR production is converted to wild-type TTR from the allograft [42•]. This approach is limited due to numerous factors including the scarcity of organs and the morbidity and mortality associated with organ transplantation, especially in more debilitated patients. The best outcomes were reported in patients with p.Val50Met TTR mutation, with early onset and short duration of symptomatic disease before transplantation [37]. Following transplantation, pre-existing variant ATTR deposits may serve as a scaffold upon which wtATTR can continue to deposit in some patients, allowing symptoms to progress [42•]. Additionally, domino liver transplant recipients may develop acquired hATTR-PN from variant TTR produced by allograft [43]. Gene silencers have been used in patients who developed progression of hATTR after liver transplantation [44, 45]. With the advent of new more effective therapies, the use of liver transplantation for hATTR has significantly declined [36].
Treatment of ATTR cardiomyopathy
As described previously, almost all mutations of the hereditary form of transthyretin amyloid cardiomyopathy (hATTR-CM) cause progressive heart failure due to accumulation of dissociated fibrils in the myocardium, resulting in heart failure symptoms and arrythmias. In addition to diuretics for symptomatic treatment and reducing afterload, the only treatment option for ATTR-CM that is approved by the FDA is the TTR stabilizer tafamidis. Prior to its approval in 2019, the only other treatment for ATTR-CM was liver transplantation to halt production of misfolded TTR or in certain cases, heart or liver/heart transplantation [46]. Additionally, another TTR stabilizer diflunisal has been available off-label, but its use was limited by potential side effects. Though first studied in patients with hATTR-PN, several small studies have shown improved survival in patients with ATTR-CM [47,48,49]. However, long-term use of diflunisal is associated with risks of nephrotoxicity and gastrointestinal adverse effects, including bleeding and perforation. Tafamidis binds to the thyroxine site of TTR, inhibiting the dissociation of tetramers into monomers and stabilizing TTR. In 2018, the results of ATTR-ACT, a multicenter randomized controlled trial on the TTR stabilizer tafamidis, showed decrease in all-cause mortality with hazard ratio of 0.70, lower rate of cardiovascular-related hospitalizations with relative risk ratio of 0.68, and lower decline in functional capacity in patients with ATTR-CM (NCT01994889) [50••]. Tafamidis was subsequently approved by the FDA in 2019 for treatment of ATTR-CM (both wtATTR and hATTR). The higher dose of tafamidis in the ATTR-ACT study was more effective than the lower dose previously used in ATTR-PN (80 mg vs 20 mg in low-dose group) [8, 50••]. Long-term follow-up showed an improved survival in patients treated with tafamidis with hazard ratio of 0.59 after approximately 58.5 months of median follow-up [51]. Unfortunately, at an initial list price of $225,000 per year, it was the most expensive cardiac drug available, limiting its affordability and access to care, and its availability for treatment of cardiomyopathy varies across the world [52, 53]. The newest TTR stabilizer studied is acoramidis (AG10) [54], and results of the ATTRibute-CM trial (NCT03860935) were recently presented at the European Society of Cardiology Congress 2023 [55]. This study found that treatment with acoramidis reduced relative risk of cardiovascular mortality by 30% and of cardiac hospitalization by 50%. Though results are promising, it is not approved by the FDA at this time.
In summary, our treatment armamentarium for ATTR-CM has increased over the last 5 years, but current available treatments remain limited due to cost and/or side effects.
Treatment of mixed phenotype
When treating multisystemic diseases, multidisciplinary approach to management is essential to improve the outcomes. Two major specialties needed by most ATTR patients are cardiology and neurology. Depending on other clinical manifestations, these patients may also benefit from evaluations by nephrologists, pulmonologists, gastroenterologists, and ophthalmologists [56, 57]. While hATTR (depending on the mutation) may have dominant cardiac or less commonly neuropathic phenotype, many of the hATTR patients have at least some features of both cardiomyopathy and neuropathy. In contrast, in wtATTR, cardiomyopathy is almost universally the dominant clinical feature, with some patients also having a mild neuropathy. Currently in the USA, approved treatment options for ATTR include tafamidis for ATTR cardiomyopathy (hATTR and wtATTR) and gene silencers (patisiran, inotersen, vutrisiran) for ATTR neuropathy (hATTR only) (Table 1) [9••, 10••, 50••]. The use of another TTR stabilizer diflunisal is limited by potential side effects including renal injury and hypertension [47]. TTR stabilizers and gene silencers have shown some beneficial effects in treatments of both hATTR cardiomyopathy and neuropathy, but the reported benefits of tafamidis for hATTR-PN and patisiran for ATTR-CM have been considered insufficient to warrant the FDA approval at this time [8, 58]. The combination of TTR stabilizers and gene silencers may potentially offer greater benefit than either medication class alone, but further studies are required. The major argument against such combination therapy is the cost. While there are no major interactions between TTR stabilizers and gene silencers, TTR stabilizers may also increase serum prealbumin (transthyretin) level although impact of these lab findings remains unclear.
Symptomatic management of dysautonomia is important for patients with mixed phenotype. Particularly, treatment of orthostatic hypotension is more complicated in heart failure patients who may not tolerate increased fluid and salt intake and may develop supine hypertension as a result of use of medications like midodrine [59].
Treatments in development and off-label therapy
Gene silencers
Eplontersen is an investigational GalNAc-conjugated ASO. The NEURO-TTRansform trial was a recent multicenter, open-label, single-group, phase 3 clinical trial that evaluated the safety and efficacy of eplontersen and compared it to a historical placebo group from the NEURO-TTR trial of inotersen [60]. Adult patients aged 18 to 82 years with FAP stages I or II were eligible to participate. Patients previously or currently treated with other TTR silencers were excluded [61]. One hundred and forty-four patients received eplontersen 45 mg subcutaneously every 4 weeks. Three primary endpoints were measured at weeks 65 or 66, including changes from baseline in serum TTR, mNIS + 7, and Norfolk QOL-DN scales. The study found an adjusted mean percentage reduction of − 81.7% in serum TTR in the eplontersen group compared to − 11.2% in the placebo group. The mean change in mNIS + 7 was 0.3 in the eplontersen group and 25.1 in the placebo group, indicating disease stability in those treated with eplontersen. The mean change in Norfolk QoL-DN was − 5.5 in the eplontersen group and 14.2 in the placebo group, suggesting a slight improvement in quality of life in patients who received eplontersen. Three patients (2.1%) on eplontersen had thrombocytopenia, but all cases were mild, and not associated with hemorrhage, without treatment interruptions or dosing changes. There were no cases of glomerulonephritis in the treatment group. Injection site reactions were observed in 8% of patients on eplontersen and 12% of patients on placebo. There were no serious adverse effects or deaths related to the study drug [60]. The main limitations of the study were its open-label design and use of a historical placebo group.
At the time of this manuscript submission, eplontersen is under the review by the Food and Drug Administration, and the decision is expected in late December 2023. If approved, eplontersen would offer an additional subcutaneous treatment option for patients with FAP, with the potential for added benefit of home self-administration of caregiver administration.
Stabilizers (diflunisal, acoramidis)
Diflunisal, a generic non-steroidal anti-inflammatory medication approved for the use of arthritis-related pain, has data showing benefits in hATTR-PN. Diflunisal stabilizes TTR tetramers, similarly like tafamidis [62]. In a randomized 1:1 controlled trial of 130 patients treated with oral diflunisal 250 mg twice daily versus placebo, the NIS + 7 showed an 18.0-point (95% CI, 9.9–26.2) difference favoring diflunisal at 2 years [47]. Quality of life based on the physical component of Short-Form Health Survey (SF-36) score also stabilized contrasting the deterioration seen in the placebo group. Gastrointestinal, renal, cardiac, and hematologic adverse events did not differ between the 2 groups. Similarly, there was no significant difference in drug-related adverse events. A long-term study of the safety and efficacy of diflunisal demonstrated that the medication is well tolerated by most patients with a sustained effect beyond 2 years [63]. Additional studies have reported the benefits for autonomic dysfunction in p.V50M FAP and in wtATTR cardiomyopathy [64, 65]. The European consensus for diagnosis, management, and treatment of hATTR-PN recommended the use of diflunisal for patients with FAP stage II [66]. These guidelines, however, preceded the approval of TTR silencers. The use of diflunisal for hATTR-PN and ATTR-CM in the USA and other countries remains off label.
Another investigational stabilizer, acoramidis (previously known as AG10), showed promise for treatment of ATTR-CM [54], but the clinical study for hATTR-PN was withdrawn by the sponsor (NCT04418024).
Gene editing therapy
The success of gene editing and/or replacement therapy in other monogenic neuromuscular disorders such as spinal muscular atrophy and Duchenne muscular dystrophy suggests hATTR should be a good candidate for this treatment modality. NTLA-2001 is an in vivo gene editing therapeutic agent based on the clustered regularly interspaced short palindromic repeats and associated Cas9 endonuclease (CRISPR-Cas9) system designed to reduce the production of both variant and wild-type TTR. It consists of a single guide RNA targeting TTR and Cas9 mRNA sequence encapsulated in a lipid nanoparticle [67•]. Sustained knockout of TTR was previously observed in pre-clinical studies [68]. The interim results of a phase 1, open-label, multicenter study assessing the safety and pharmacodynamic effects of two different doses of NTLA-2001 (0.1 mg kg and 0.3 mg/kg) were published in 2021. Six adult patients with hATTR-PN received a single dose of NTLA-2001 intravenously with a dose-dependent reduction of serum TTR that was observed at days 14 and 28 (mean 52% in the lower dose group and 87% in the higher dose group at day 28). All patients completed the infusion without interruptions. Three patients experienced mild adverse effects during or after the infusion. One patient had an infusion-related reaction. Studies in patients with hATTR-PN and hATTR-CM are ongoing with expected completion in 2026 (NCT04601051).
Amyloid removal agents
Currently available treatments for ATTR reduce or block the formation of tissue amyloid deposits, and various strategies may be considered to remove existing amyloid plaques including anti-amyloid antibodies and doxycycline with tauroursodeoxycholic acid. At this time, there are no approved treatments to remove ATTR amyloid, but recent approvals of monoclonal antibodies targeting amyloid plaques for treatment of Alzheimer’s disease suggest that such strategy may be effective [69]. Some of treatments that are not specific for removal of ATTR deposits have been used as potential treatment for other types of amyloidoses as well (e.g., doxycycline in AL). Treatment with doxycycline and tauroursodeoxycholic acid leads to improvement of left ventricular strain in 38% of patients with ATTR-CM treated for a median time of 22 months [70]. A recent phase 1 study of an investigational anti-ATTR antibody NI006 demonstrated reduction of amyloid on imaging study (cardiac MRI, scintigraphy), and the reduction of NT-proBNP and troponin-T levels contrasting deterioration in placebo-treated group (NCT04360434) [71]. Another investigational antibody targeting an epitope on misfolded and aggregated TTR, NNC6019, is now in a phase 2 clinical study for treatment of ATTR-CM with planned completion in early 2025 (NCT05442047) [72]. Additionally, there was a recent report of immune response leading to spontaneous clearance of cardiac amyloid deposits in 3 patients attributed to the immune response in the presence of a high titer of circulating polyclonal IgG antibodies reactive to human ATTR [73•]. A novel approach using chimeric antigen receptor-engineered macrophages (CAR-M) is currently at a preclinical stage [74].
Conclusions and future directions
Currently available treatments have changed natural history of hATTR-PN and ATTR-CM with slowing of progression, reduced hospitalization, and improved mortality. However, while current gene silencers are effective in targeting and knocking down the production of TTR in the liver [38], they do not block TTR production in the eye and CNS, similarly as what happens after liver transplantation. Liver transplantation was the first treatment for hATTR, before the advent of TTR stabilizers and gene silencing as it stopped the production of variant TTR in the liver [42•]. However, some of p.V50M hATTR patients who underwent liver transplantation developed transient neurologic events attributable to CNS ATTR at 8 to 17 years after the transplantation [75, 76]. Therefore, we may expect the onset of CNS ATTR in some hATTR-PN patients after long-term treatment and prolonged survival with gene silencers. A potential role for tafamidis and tolcapone in treatment of CNS hATTR may be considered as well as both drugs penetrate CNS with potentially effective CSF concentrations [77, 78], although individual case reports do not support use of tafamidis in OLMA [79].
Additional potential treatments for hATTR that may get approved in the near future by drug control agencies include a new ASO gene silencer eplontersen for treatment of hATTR neuropathy, and TTR stabilizer acoramidis for treatment of ATTR cardiomyopathy [60, 80]. Along with approval for hATTR-PN, vutrisiran may also get approved for cardiomyopathy.
Management of ATTR has rapidly evolved since tafamidis was approved for treatment of hATTR-associated polyneuropathy in Europe in 2011, and currently in the USA there are 4 medications approved for treatment of ATTR with more to come. Future management protocols may include combinations of gene silencers, TTR stabilizers, and amyloid removal agents, but the cost may become the limiting factor. The role of Cas9-CRISPR in treatment of hATTR and other hereditary disorders is being investigated as well [67•].
As management of ATTR continues to evolve, some of the important challenges include [1] developing effective treatment of ATTR in the CNS and the eye; [2] removal of existing ATTR deposits in different organs and tissues; and [3] optimization of long-term gene silencing (optimal percentage of TTR suppression, frequency and route of dosing, effects of long-term prealbumin knockdown).
Availability of Data and Materials
No datasets were generated or analyzed during the current study.
Abbreviations
- AL:
-
Light-chain amyloidosis
- ASO:
-
Antisense oligonucleotide
- ATTR:
-
Transthyretin amyloidosis
- ATTR-CM:
-
Transthyretin amyloid cardiomyopathy
- CNS:
-
Central nervous system
- CTS:
-
Carpal tunnel syndrome
- FAP:
-
Familial amyloid polyneuropathy
- GalNAc:
-
N-Acetylgalactosamine
- hATTR:
-
Hereditary transthyretin amyloidosis
- hATTR-CM:
-
Hereditary transthyretin amyloid cardiomyopathy
- hATTR-PN:
-
Hereditary transthyretin amyloid peripheral neuropathy
- mRNA:
-
Messenger RNA
- Norfolk QOL-DN:
-
Norfolk Quality of Life-Diabetic Neuropathy
- OLMA:
-
Oculoleptomeningeal amyloidosis
- PND:
-
Polyneuropathy Disability scale
- RISC:
-
RNA-induced silencing complex
- siRNA:
-
Small interfering RNA
- TTR:
-
Transthyretin amyloidosis
- wtATTR:
-
Wild-type transthyretin amyloidosis
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Obici L, Adams D. Acquired and inherited amyloidosis: knowledge driving patients’ care. J Peripher Nerv Syst. 2020;25(2):85–101.
Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036–43.
Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–20.
Wininger AE, Phelps BM, Le JT, Harris JD, Trachtenberg BH, Liberman SR. Musculoskeletal pathology as an early warning sign of systemic amyloidosis: a systematic review of amyloid deposition and orthopedic surgery. BMC Musculoskelet Disord. 2021;22(1):51.
Wajnsztajn Yungher F, Kim A, Boehme A, Kleyman I, Weimer LH, Maurer MS, et al. Peripheral neuropathy symptoms in wild type transthyretin amyloidosis. J Peripher Nerv Syst. 2020;25(3):265–72.
Zivkovic S, Soman P, Lacomis D. Late-onset peripheral neuropathy in patients with wild type transthyretin amyloidosis (wtATTR). Amyloid. 2020;27(2):142–3.
Papagianni A, Ihne S, Zeller D, Morbach C, Uceyler N, Sommer C. Clinical and apparative investigation of large and small nerve fiber impairment in mixed cohort of ATTR-amyloidosis: impact on patient management and new insights in wild-type. Amyloid. 2022;29(1):14–22.
Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92.
•• Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. Pivotal study showing efficacy of gene silencing of TTR with patisiran.
•• Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. Pivotal study showing efficacy of gene silencing of TTR with inotersen.
Kaku MC, Bhadola S, Berk JL, Sanchorawala V, Connors LH, Lau KHV. Neurological manifestations of hereditary transthyretin amyloidosis: a focus on diagnostic delays. Amyloid. 2022;29(3):184–9.
Coelho T, Dispenzieri A, Grogan M, Conceicao I, Waddington-Cruz M, Kristen AV, et al. Patients with transthyretin amyloidosis enrolled in THAOS between 2018 and 2021 continue to experience substantial diagnostic delay. Amyloid. 2023:1–4. online ahead of print
Wang AK, Fealey RD, Gehrking TL, Low PA. Patterns of neuropathy and autonomic failure in patients with amyloidosis. Mayo Clin Proc. 2008;83(11):1226–30.
Misu K, Hattori N, Nagamatsu M, Ikeda S, Ando Y, Nakazato M, et al. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features Brain. 1999;122(Pt 10):1951–62.
Conceicao I, Gonzalez-Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.
Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy: a systematic review. JAMA. 2020;324(1):79–89.
Hens D, Wyers L, Claeys KG. Validation of an artificial intelligence driven framework to automatically detect red flag symptoms in screening for rare diseases in electronic health records: hereditary transthyretin amyloidosis polyneuropathy as a key example. J Peripher Nerv Syst. 2023;28(1):79–85.
•• Adams D, Coelho T, Obici L, Merlini G, Mincheva Z, Suanprasert N, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675–82. Important study demonstrating natural history of hATTR-PN with rapid progression.
Lozeron P, Mariani LL, Dodet P, Beaudonnet G, Theaudin M, Adam C, et al. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology. 2018;91(2):e143–52.
Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040–50.
Milandri A, Farioli A, Gagliardi C, Longhi S, Salvi F, Curti S, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22(3):507–15.
Karam C, Dimitrova D, Christ M, Heitner SB. Carpal tunnel syndrome and associated symptoms as first manifestation of hATTR amyloidosis. Neurol Clin Pract. 2019;9(4):309–13.
Salvalaggio A, Coraci D, Cacciavillani M, Obici L, Mazzeo A, Luigetti M, et al. Nerve ultrasound in hereditary transthyretin amyloidosis: red flags and possible progression biomarkers. J Neurol. 2021;268(1):189–98.
Poncelet A, Weiler M, Hegenbart U, Sam G, Schonland S, Purrucker JC, et al. Dual-echo turbo spin echo and 12-echo multi spin echo sequences as equivalent techniques for obtaining T2-relaxometry data: application in symptomatic and asymptomatic hereditary transthyretin amyloidosis as a surrogate disease. Invest Radiol. 2022;57(5):301–7.
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12.
Wlodarski R, Seibert K, Issa NP, O’Brien-Penney B, Soliven B, Sarswat N, et al. (99m) Technetium-pyrophosphate bone scan: a potential biomarker for the burden of transthyretin amyloidosis in skeletal muscle: a preliminary study. Muscle Nerve. 2023;67(2):111–6.
Shouman K, Broski SM, Muchtar E, Pendleton CA, Johnson GB, Tracy J, et al. Novel imaging techniques using (18) F-florbetapir PET/MRI can guide fascicular nerve biopsy in amyloid multiple mononeuropathy. Muscle Nerve. 2021;63(1):104–8.
•• Sousa L, Coelho T, Taipa R. CNS involvement in hereditary transthyretin amyloidosis. Neurology. 2021;97(24):1111–9. Important overview of CNS involvement in hATTR.
Adams D, Ando Y, Beirao JM, Coelho T, Gertz MA, Gillmore JD, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–22.
Benson MD, Berk JL, Dispenzieri A, Damy T, Gillmore JD, Hazenberg BP, et al. Tissue biopsy for the diagnosis of amyloidosis: experience from some centres. Amyloid. 2022;29(1):8–13.
Hill MM, Dasari S, Mollee P, Merlini G, Costello CE, Hazenberg BPC, et al. The clinical impact of proteomics in amyloid typing. Mayo Clin Proc. 2021;96(5):1122–7.
Freeman R, Gonzalez-Duarte A, Barroso F, Campagnolo M, Rajan S, Garcia J, et al. Cutaneous amyloid is a biomarker in early ATTRv neuropathy and progresses across disease stages. Ann Clin Transl Neurol. 2022;9(9):1370–83.
Leonardi L, Adam C, Beaudonnet G, Beauvais D, Cauquil C, Not A, et al. Skin amyloid deposits and nerve fiber loss as markers of neuropathy onset and progression in hereditary transthyretin amyloidosis. Eur J Neurol. 2022;29(5):1477–87.
Obici L, Kuks JB, Buades J, Adams D, Suhr OB, Coelho T, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29 Suppl 1(Suppl 1):S27–35.
• Quan D, Obici L, Berk JL, Ando Y, Aldinc E, White MT, et al. Impact of baseline polyneuropathy severity on patisiran treatment outcomes in the APOLLO trial. Amyloid. 2023;30(1):49–58. Study demonsrating benefits of early treatment of hATTR-PN for preservation of function.
Ando Y, Adams D, Benson MD, Berk JL, Plante-Bordeneuve V, Coelho T, et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022;29(3):143–55.
Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–54.
Brannagan TH 3rd, Berk JL, Gillmore JD, Maurer MS, Waddington-Cruz M, Fontana M, et al. Liver-directed drugs for transthyretin-mediated amyloidosis. J Peripher Nerv Syst. 2022;27(4):228–37.
Dyck PJB, Gonzalez-Duarte A, Obici L, Polydefkis M, Wiesman JF, Antonino I, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. 2019;405: 116424.
Tegsedi REMS (Risk Evaluation and Mitigation Strategy) Program. [Available from: https://www.tegsedirems.com/.
•• Adams D, Tournev IL, Taylor MS, Coelho T, Plante-Bordeneuve V, Berk JL, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023;30(1):1–9. Phase 3 study demonstrating efficacy of vutrisiran for treatment of hATTR-PN.
• Benson MD. Liver transplantation and transthyretin amyloidosis. Muscle Nerve. 2013;47(2):157–62. Important overview of liver transplantation in hATTR.
Mnatsakanova D, Zivkovic SA. Iatrogenic amyloid polyneuropathy after domino liver transplantation. World J Hepatol. 2017;9(3):126–30.
Moshe-Lilie O, Dimitrova D, Heitner SB, Brannagan TH 3rd, Zivkovic S, Hanna M, et al. TTR gene silencing therapy in post liver transplant hereditary ATTR amyloidosis patients. Amyloid. 2020;27(4):250–3.
Schmidt HH, Wixner J, Plante-Bordeneuve V, Munoz-Beamud F, Llado L, Gillmore JD, et al. Patisiran treatment in patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy after liver transplantation. Am J Transplant. 2022;22(6):1646–57.
Razvi Y, Porcari A, Di Nora C, Patel RK, Ioannou A, Rauf MU, et al. Cardiac transplantation in transthyretin amyloid cardiomyopathy: outcomes from three decades of tertiary center experience. Front Cardiovasc Med. 2022;9:1075806.
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–67.
Ibrahim M, Saint Croix GR, Lacy S, Fattouh M, Barillas-Lara MI, Behrooz L, et al. The use of diflunisal for transthyretin cardiac amyloidosis: a review. Heart Fail Rev. 2022;27(2):517–24.
Tschope C, Elsanhoury A. Treatment of transthyretin amyloid cardiomyopathy: the current options, the future, and the challenges. J Clin Med. 2022;11(8).
•• Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16. pivotal study demonstrating benefits of tafamidis for treatment of ATTR-CM.
Elliott P, Drachman BM, Gottlieb SS, Hoffman JE, Hummel SL, Lenihan DJ, et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail. 2022;15(1): e008193.
Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–24.
Wardhere A, Bampatsias D, Fine N, Garcia-Pavia P, Grogan M, Kristen AV, et al. Heterogeneous worldwide access and pricing of tafamidis. Amyloid. 2023:1–3. (online ahead of print)
Masri A, Aras M, Falk RH, Grogan M, Jacoby D, Judge DP, et al. Long-term safety and tolerability of acoramidis (AG10) in symptomatic transthyretin amyloid cardiomyopathy: updated analysis from an ongoing phase 2 open-label extension study. ISA 2022; Heidelberg, Germany, 2022. p. 47. (abstract)
Gillmore JD, Judge DP, Cappelli F, Fontana M, al. e. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. ESC 2023; Amsterdam, Netherlands, 2023. (abstract)
Bumma N, Kahwash R, Parikh SV, Isfort M, Freimer M, Vallakati A, et al. Multidisciplinary amyloidosis care in the era of personalized medicine. Front Neurol. 2022;13: 935936.
Obici L, Callaghan R, Ablett J, Bibiloni C, Bueser T, Conceicao I, et al. Consensus recommendations on holistic care in hereditary ATTR amyloidosis: an international Delphi survey of patient advocates and multidisciplinary healthcare professionals. BMJ Open. 2023;13(9): e073130.
Maurer MS, Kale P, Fontana M, Berk JL, Grogan M, Gustafsson F, et al. Patisiran treatment in patients with transthyretin cardiac amyloidosis. N Engl J Med. 2023;389(17):1553–65.
Olshansky B, Muldowney J. Cardiovascular safety considerations in the treatment of neurogenic orthostatic hypotension. Am J Cardiol. 2020;125(10):1582–93.
Coelho T, Marques W Jr, Dasgupta NR, Chao CC, Parman Y, Franca MC Jr, et al. Eplontersen for hereditary transthyretin amyloidosis with polyneuropathy. JAMA. 2023;330(15):1448–58.
Coelho T, Waddington Cruz M, Chao CC, Parman Y, Wixner J, Weiler M, et al. Characteristics of patients with hereditary transthyretin amyloidosis-polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an open-label phase 3 study of eplontersen. Neurol Ther. 2023;12(1):267–87.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–49.
Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79–83.
Siddiqi OK, Mints YY, Berk JL, Connors L, Doros G, Gopal DM, et al. Diflunisal treatment is associated with improved survival for patients with early stage wild-type transthyretin (ATTR) amyloid cardiomyopathy: the Boston University Amyloidosis Center experience. Amyloid. 2022;29(2):71–8.
Takahashi R, Ono K, Shibata S, Nakamura K, Komatsu J, Ikeda Y, et al. Efficacy of diflunisal on autonomic dysfunction of late-onset familial amyloid polyneuropathy (TTR Val30Met) in a Japanese endemic area. J Neurol Sci. 2014;345(1–2):231–5.
Adams D, Suhr OB, Hund E, Obici L, Tournev I, Campistol JM, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29 Suppl 1(Suppl 1):S14–26.
• Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. Important study demonstrating potential benefits of gene editing as a treatment of hATTR.
Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22(9):2227–35.
Cummings J, Osse AML, Cammann D, Powell J, Chen J. Anti-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. BioDrugs. 2023. online ahead of print.
Karlstedt E, Jimenez-Zepeda V, Howlett JG, White JA, Fine NM. Clinical experience with the use of doxycycline and ursodeoxycholic acid for the treatment of transthyretin cardiac amyloidosis. J Card Fail. 2019;25(3):147–53.
Garcia-Pavia P, Aus dem Siepen F, Donal E, Lairez O, van der Meer P, Kristen AV, et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N Engl J Med. 2023;389(3):239–50.
Maurer MS, Buchholtz K, Engelman MM, Grogan M, Hovingh GK, Kristen AV, et al. NNC6019–0001, a humanized monoclonal antibody, in patients with transthyretin amyloid cardiomyopathy (ATTR-CM): rationale and study design of a phase 2, randomized, placebo-controlled trial. ISA 2022; Heidelberg, Germany, 2022. p. 746–7. abstract
• Fontana M, Gilbertson J, Verona G, Riefolo M, Slamova I, Leone O, et al. Antibody-associated reversal of ATTR amyloidosis-related cardiomyopathy. N Engl J Med. 2023;388(23):2199–201. Demonstration of antibody-associated removal of ATTR in the heart.
Balanchandran M, Foster S, Jackson J, Richey T, Martin E, Kennel S, et al. Development of novel human chimeric antigen receptor-macrophages (CAR-M) as a potential therapeutic for amyloid clearance. ISA 2022; Heidelberg, Germany, 2022. p. 98–9. abstract
Salvi F, Pastorelli F, Plasmati R, Morelli C, Rapezzi C, Bianchi A, et al. Brain microbleeds 12 years after orthotopic liver transplantation in Val30Met amyloidosis. J Stroke Cerebrovasc Dis. 2015;24(6):e149–51.
Sekijima Y, Yazaki M, Oguchi K, Ezawa N, Yoshinaga T, Yamada M, et al. Cerebral amyloid angiopathy in posttransplant patients with hereditary ATTR amyloidosis. Neurology. 2016;87(8):773–81.
Takahashi Y, Ohashi N, Takasone K, Yoshinaga T, Yazaki M, Roberts M, et al. CSF/plasma levels, transthyretin stabilisation and safety of multiple doses of tolcapone in subjects with hereditary ATTR amyloidosis. Amyloid. 2022;29(3):190–6.
Tsai FJ, Jaeger M, Coelho T, Powers ET, Kelly JW. Tafamidis concentration required for transthyretin stabilisation in cerebrospinal fluid. Amyloid. 2023;30(3):279–89.
Salvi F, Volpe R, Pastorelli F, Bianchi A, Vella A, Rapezzi C, et al. Failure of tafamidis to halt progression of Ala36Pro TTR oculomeningovascular amyloidosis. J Stroke Cerebrovasc Dis. 2018;27(9):e212–4.
Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74(3):285–95.
Author information
Authors and Affiliations
Contributions
All authors have participated in the drafting of the manuscript and editing. All authors reviewed the final version of the manuscript and agreed with its submission.
Corresponding author
Ethics declarations
Conflict of Interest
Sasha A. Živković — Consulting and/or advisory boards with Alnylam Pharmaceuticals, Astra Zeneca, Argenx, and Takeda. J. David Avila — Speaker for Alnylam, Argenx, Alexion, and UCB; consulting for Alnylam Pharmaceuticals and Alexion. Cesia Gallegos-Kattan — Advisory board for Alnylam Pharmaceuticals. Dianna Quan — Research funding, speaking fees, and travel expenses from Alnylam Pharmaceuticals; research funding from Pfizer, Ionis, Cytokinetics, Momenta/Janssen, Viela Bio, and Avidity Biosciences.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Živković, S.A., Avila, J.D., Gallegos-Kattan, C. et al. Update on Amyloid Polyneuropathy and Treatment. Curr Treat Options Neurol 26, 51–66 (2024). https://doi.org/10.1007/s11940-024-00780-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-024-00780-z