
Abstract
Purpose of review
The term neuromyelitis optica spectrum disorders (NMOSD) encompasses a constellation of potentially devastating inflammatory maladies of the central nervous system that predominantly affect the optic nerve(s) and spinal cord. Neurologic disability occurs as a result of severe, immune-mediated demyelination and irreversible axonal damage. Significant advances have been made in defining the clinical characteristics of NMOSD, including key examination findings and specific imaging and laboratory indicators. This review will provide the clinician with critical knowledge to allow for both early diagnosis, and consideration of available treatment strategies.
Recent findings
As attacks of NMOSD tend to be severe and irreversible, relapse prevention is essential to the mitigation of permanent neurologic disability. Newly FDA-approved monoclonal antibodies have become available for the management of NMOSD; these may provide clinical stability and reduction in relapses over time.
Summary
NMOSD frequently causes irreversible neurologic disability, and therefore early diagnosis and appropriate pharmacologic intervention are essential. Several previously established as well as newer medications are available for the treatment of acute attacks and for relapse prevention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The first clinical descriptions of neuromyelitis optica spectrum disorders (NMOSD, previously known as Devic disease or neuromyelitis optica [NMO]), emerged in the late-nineteenth century [1, 2]. Typical patients presented with a monophasic course of bilateral (or rapidly sequential) optic neuritis and myelitis. The visual and/or motor sequelae of this condition were severe, and recovery was limited or non-existent.
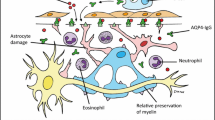
Over the course of time the presenting features, degree of recovery, presence/absence of serial attacks, and the extent of accumulated disability have proven to be variable [3]. Moreover, advances in the understanding of the pathogenesis and neuropathology of NMOSD, as well as its distinct imaging characteristics, biomarkers and treatment strategies, have allowed NMOSD to be definitively identified as distinct from multiple sclerosis (MS), and have resulted in the development of clinically useful diagnostic criteria to assist in the early identification of patients with this potentially devastating immune-mediated disease (Fig. 1).

Reproduced from: Wingerchuk DM, Banwell B, Bennett JL, Cabre P et al. Neurology 2015;85(2).
International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. AQP4-IgG, anti–aquaporin-4 immunoglobulin G; LETM, longitudinally extensive transverse myelitis; MRI, magnetic resonance imaging; NMOSD, neuromyelitis optica spectrum disorder.
The pathogenesis of NMOSD is precipitated by a complement-mediated immune attack that preferentially targets astrocytes, with the secondary effect of florid demyelination and neuronal damage that favors the optic nerves and longitudinally extensive segments of the spinal cord. This is due to the characteristic distribution of the water channel protein aquaporin-4 (AQP4) within the central nervous system [4, 5]. AQP4 is the target antigen of NMO-immunoglobulin G (IgG), also known as anti-AQP4, which plays a direct role in the pathogenesis of NMOSD [6]. Binding of NMO-IgG to AQP4 initiates a cascade of events leading to axonal loss, perivascular lymphocytic infiltration, vascular proliferation, and necrosis [7].
An NMOSD disease-specific autoantibody test is available for both serum and cerebrospinal fluid (CSF). A subset of patients with a phenotype of NMOSD may also harbor anti-myelin oligodendrocyte glycoprotein (MOG) antibodies.
The prevalence of NMOSD ranges from 0.5 to 10 per 100,000 [8,9,10], with variations based on ethnic, geographic, and gender-related factors [11, 12]. The median age of onset is 32–41 years (higher than in multiple sclerosis), and the incidence may be up to 10-times higher in women than in men [13]. The onset of NMO is most commonly sporadic, but some familial associations have been reported [14].
The principal clinical features of NMOSD are attacks of bilateral (or rapidly sequential) optic neuritis that are typically severe and result in profound visual loss, and/or longitudinally extensive transverse myelitis resulting in weakness, impaired mobility, sensory changes, and bladder dysfunction. A rare but highly characteristic constellation of symptoms including intractable nausea, emesis, and hiccups (the so-called area postrema syndrome) has been described. Other patients may experience narcolepsy, reversible posterior leukoencephalopathy syndrome, or neuroendocrine disorders. Most patients follow a relapsing course [15], although in distinction from MS, the neurologic deficits attendant to each attack have variable (and often incomplete) recovery over weeks to months [16]. Consequently, serial attacks over time result in the accumulation of residual disability with many patients becoming blind or paraplegic within five years of onset [17, 18].
Due to the great risk of irreversible and accumulated disability over time, early diagnosis of NMOSD is essential. Clinical presentations that should raise suspicion for NMOSD include [19••] the following:
-
Simultaneous or sequential bilateral optic neuritis (even if asymmetric), especially with involvement of the optic chiasm, the presence of an altitudinal visual field defect, or severe with incomplete recovery.
-
Complete spinal cord syndrome, especially with paroxysmal tonic spasms.
-
Area postrema syndrome with intractable hiccups or nausea and vomiting.
Patients presenting with the above clinical features should undergo brain and spinal cord imaging with magnetic resonance imaging (MRI), serum antibody testing for AQP4-IgG (a cell-based assay is strongly encouraged) and MOG antibody, and possibly CSF analysis. Diagnostic criteria for NMOSD have been established as summarized in Table 1 [19••], and are helpful in confirming the diagnosis in the appropriate clinical setting.
Treatment
Diet and lifestyle
-
No specific dietary modifications or supplements have been shown to be beneficial with regard to the prevention or treatment of NMOSD.
-
Patients should be advised to maintain a well-balanced diet that promotes good overall health.
-
Within the limits of the patient’s physical abilities and other health considerations, regular exercise will assist in the optimization of baseline neurologic function.
Pharmacologic treatment
-
The importance of treating both acute and recurrent attacks of NMOSD is based on the likelihood of attack-related disability, poor prognosis for post-attack recovery, and overall high risk of mortality in untreated patients [20].
-
Early recognition of acute NMOSD features is critical, particularly in individuals without an established diagnosis, as some data suggest that patients may experience greater recovery if concurrent treatment with glucocorticoids and plasma exchange is initiated within days of symptom onset (and typically before AQP4-IgG antibody results become available) [21•].
-
For acute attacks that are severe or refractory to early administration of glucocorticoids, therapeutic plasma exchange should be provided.
-
For patients with an established diagnosis of NMOSD, long-term immunotherapy is recommended to help prevent recurrent attacks and accumulated disability. Immunosuppression is usually continued for at least five years for patients who are seropositive for AQP4-IgG due to the high risk of relapse.
-
While there is no difference in the management of seronegative vs seropositive NMOSD, newer medications such as eculizumab and inebilizumab are approved only for patients who are seropositive for AQP4-IgG (see below).
Acute attacks
Methylprednisolone (IVMP)
As soon as practicable following onset of symptoms, patients with known or suspected exacerbations of NMOSD should receive intravenous methylprednisolone (IVMP) at a dose of 1000 mg daily for three to five consecutive days, similar to the regimen established for the treatment of acute attacks of MS [20]. Contraindications to this treatment include the presence of a concomitant systemic infection, and caution should be exercised in patients with uncontrolled diabetes. However, given the short duration of therapy and the expectation that most patients will receive treatment in the inpatient setting, complications related to this treatment are few. Potential interactions with macrolide antibiotics (which may cause a decrease in corticosteroid clearance), warfarin (whose response may be inhibited by corticosteroids), and non-steroidal anti-inflammatory agents (which may increase the risk for gastrointestinal side effects) should be considered. Principal side effects during a three-to-five-day course of treatment may include increased appetite, increased energy level, insomnia, and irritability. The cost for a five-day course of therapy is approximately $350, not including the cost of infusion and other ancillary services.
Therapeutic plasma exchange (TPE)
For patients with severe attacks, or symptoms that are refractory to methylprednisolone, a five-to-seven day course of plasma exchange is recommended [22•, Class I). Most common protocols allow for 1 to 1.5 plasma volumes to be exchanged per procedure, which are performed on an every-other-day basis. Potential complications of this therapy are related to central venous access, anticoagulation and replacement fluids, as well as hypocalcemia or alterations in acid–base homeostasis. As TPE is an invasive procedure, patients must be admitted to hospital. TPE is an expensive therapy, with some cost estimates in the range of $100,000 per course of treatment.
Relapse prevention: FDA-approved medications
Eculizumab
Eculizumab is a humanized monoclonal antibody that acts as a complement inhibitor, binding to C5 and inhibiting the formation of C5b-induced membrane attack complex (MAC). A randomized controlled-trial in AQP4-IgG seropositive patients achieved its primary endpoint of delaying the first adjudicated NMOSD relapse (the annualized relapse rates for the eculizumab and placebo groups were 0.02 and 0.35 respectively (absolute risk reduction [ARR] 33 percent, rate ratio 0.04, 95% CI 0.01–0.15)) and that the therapy was well-tolerated [23••, Class I]. Eculizumab is administered as an IV infusion at a dose of 900 mg weekly for four doses, followed by 1200 mg every-two-weeks thereafter as a maintenance dose. Commonly reported side effects were mild and included headache, upper respiratory tract infections, and contusions. Eculizumab may be associated with an increased risk of Neisseria meningitidis infection, and patients must be immunized with meningococcal vaccines prior to initiating therapy (or receive concurrent antimicrobial prophylaxis if acute treatment is indicated). As a result of this, eculizumab is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. Approval for this medication requires AQP4-IgG seropositivity. At an estimated list price of $400,000 per year, eculizumab is an expensive treatment, although robust cost reduction strategies are available to assist patients.
Inebilizumab
Inebilizumab is a humanized monoclonal antibody that depletes lymphocytes through binding to the CD19 surface antigen of B cells. A randomized clinical trial comparing inebilizumab to placebo [24••, Class I] was ended at the 6.5 month interim analysis after it was recognized that patients treated with the medication had significantly fewer attacks as compared to patients assigned to placebo. Therefore, this phase 3 clinical trial achieved its primary endpoint of delaying the time-to-first-relapse in patients on therapy (12 versus 39 percent, ARR 27 percent, hazard ratio [HR] 0.27, 95% CI 0.15–0.50). Inebilizumab is administered by IV infusion by two 300 mg doses given two weeks apart, followed by a maintenance infusion of 300 mg every-six-months thereafter. Prior to each infusion, premedication with a glucocorticoid, an antihistamine, and an antipyretic is required, and patients must be carefully monitored throughout for the emergence of infusion reactions. Common adverse reactions in the randomized trial included urinary tract infection, headache, arthralgia, nausea, and back pain. Contraindications to therapy include active infection (scheduled infusions must be delayed until the infection resolves), and patients must be pre-screened for hepatitis B virus, quantitative serum immunoglobulins, and tuberculosis prior to the first dose. Moreover, immunoglobin levels must be monitored during the course of treatment, and after discontinuation of therapy, until B cell repletion has been achieved. Accordingly, vaccination with live or live-attenuated vaccines is not recommended while taking inebilizumab. Approval for this medication requires AQP4-IgG seropositivity. Annual pricing estimates are in the range of $200,000 to $280,000.
Satrilizumab
Satralizumab is a humanized monoclonal antibody that suppresses inflammation through binding interleukin-6 (IL-6) receptors thereby blocking subsequent IL-6 signaling pathways. One randomized controlled trial [25••, Class I] demonstrated fewer and less frequent relapses in both AQP4-IgG seropositive and seronegative patients, thereby broadening the clinical setting in which this treatment has demonstrated efficacy. A second trial which excluded patients on concomitant immunosuppressant therapy also demonstrated a reduction in the rate of NMOSD relapses [26••, Class I]. Both of these trials achieved their endpoints of reducing the risk of subsequent NMOSD attacks, between 51 and 66% as compared to placebo. Administration of satrilizumab (based on the design of the clinical trials) is via subcutaneous injection of 120 mg at weeks 0, 2, and 4 followed by subcutaneous maintenance therapy of satrilizumab 120 mg every-four-weeks thereafter. Based on data from the clinical trials, the most common adverse reactions included headache, nasopharyngitis, upper respiratory tract infection, gastritis, fatigue, and nausea. Overall, however, satrilizumab was generally well-tolerated. A 1-year course of treatment is estimated to cost approximately $190,000.
Relapse prevention: other therapies
Rituximab
Rituximab is a humanized monoclonal antibody that binds to the CD20 antigen of B cell lymphocytes and Fc receptors, resulting in pharmacologically significant B cell depletion. Although this is a commonly used immunotherapy for NMOSD, it is technically “off-label” and therefore does not require AQP4-IgG seropositivity. In small, open-label randomized trials comparing rituximab to azathioprine [27] and placebo [28], reductions in annualized relapse rate were reported. In assessing for clinical effect, patients were provided with IV rituximab at a dose of 375 mg/m2 once-weekly for four weeks, followed by a maintenance dose of 2000 mg (given as two 1000 mg doses separated by two weeks) at 24 and 48 weeks. Principal adverse effects include infusion reactions, hepatitis B reactivation, and progressive multifocal leukoencephalopathy (PML). Relative to other monoclonal antibody therapies, the cost of rituximab is less expensive at approximately $36,000 per year.
Tocilizumab
Tocilizumab is a monoclonal antibody that suppresses inflammation through binding interleukin-6 (IL-6) receptors thereby blocking subsequent IL-6 signaling pathways. It is not FDA-approved for the treatment of NMOSD, and therefore does not require AQP4-IgG seropositivity for administration. Tocilizumab is given at a dose of 8 mg/kg IV on an every-four-week basis. It has been shown in trials to be superior to azathioprine in reducing the relapse rate [29], and to be associated with clinical stabilization or improvement in patients with NMOSD that had not responded to other immunosuppressive treatments [30]. Patients treated with tocilizumab may be at increased risk for serious infections including tuberculosis, invasive fungal infections, and opportunistic bacterial infections and therefore close clinical monitoring is required. Tocilizumab is not recommended for patients with elevated liver function tests, and laboratory monitoring is required to detect abnormalities in neutrophils, platelets, and lipids. Estimated overall costs of infusions per year are approximately $36,000.
Other considerations
Observational studies regarding other systemic immunotherapeutic agents have suggested some efficacy from azathioprine and mycophenolate mofetil (120, 154, 155, Class IV) in the treatment of NMOSD. Limited observational evidence suggests that treatment of NMOSD with interferon beta, natalizumab, or fingolimod is not effective and may be harmful [31,32,33,34, Class IV].
Assistive devices
-
For patients with visual loss as a result of unilateral or bilateral optic neuritis, consultation with a low-vision specialist may assist in identifying technology devices and/or adaptive strategies to enhance quality-of-life.
-
Mobility aids should be provided for patients with limitations following transverse myelitis.
Physical/speech therapy and exercise
-
Due to the prevalence of severe vision loss and/or loss of mobility as a result of transverse myelitis in patients with NMO, early initiation of physical therapy and exercise therapy may be of benefit.
-
Patients with NMO have showed functional improvements in both Expanded Disability Status Scale (EDSS) and other functional measures similar to patients with multiple sclerosis [35].
Pediatric considerations
Children
Although firm conclusions are limited by small numbers of patients, the available data suggest that a substantial minority of children with NMOSD have brain involvement at presentation associated with clinical features of encephalopathy, seizures, and/or lesions on brain MRI resembling those typically seen with multiple sclerosis or acute disseminated encephalomyelitis [36, 37].
Conclusion
Neuromyelitis optica spectrum disorder has now been identified as a distinct pathophysiologic entity, and the recent establishment of clinical criteria and a unique serum biomarker has allowed for early and accurate diagnosis in the appropriate setting. Practitioners should be aware of the specific presenting features of NMOSD, and be familiar with the most effective method for confirming the diagnosis, as delays in acute treatment followed by initiation of relapse prevention can result in devastating and irreversible neurologic disability. Fortunately, newer FDA-approved medications are showing promise in stabilizing the disease process.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
Devic E. Myélite aiguë compliquée de névrite optique. Bull Med (Paris). 1894;8:1033.
Gault F. De la neuromyélite optique aiguë, Lyon 1894.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15. https://doi.org/10.1016/S1474-4422(07)70216-8.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. https://doi.org/10.1084/jem.20050304.
Jung JS, Bhat RV, Preston GM, Guggino WB, Barbaran JM, Agre P. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci USA. 1994;91:13052–6. https://doi.org/10.1073/pnas.91.26.13052.
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12. https://doi.org/10.1016/S0140-6736(04)17551-X.
Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11:535–44. https://doi.org/10.1016/S1474-4422(12)70133-3.
Flanagan EP, Cabre P, Weinshenker BG, St Sauver J, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79(5):775–83. https://doi.org/10.1002/ana.24617.
Bukhari W, Prain KM, Waters P, Woodhall M, O’Gorman CM, Clarke L, et al. Incidence and prevalence of NMOSD in Australia and New Zealand. J Neurol Neurosurg Psychiatry. 2017;88:632–8. https://doi.org/10.1136/jnnp-2016-314839.
Miyamoto K, Fujihara K, Kira JI, Kuriyama N, Matsui M, Tamakoshi A, et al. Nationwide epidemiological study of neuromyelitis optica in Japan. J Neurol Neurosurg Psychiatry. 2018;89:667–8. https://doi.org/10.1136/jnnp-2017-317321.
Mori M, Kuwabara S, Paul F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89:555–6. https://doi.org/10.1136/jnnp-2017-317566.
Kim SH, Mealy MA, Levy M, Schmidt F, Ruprecht K, Paul F, et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology. 2018;91(22):e2089. https://doi.org/10.1212/WNL.0000000000006574.
Wingerchuk DM. Neuromyelitis optica: effect of gender. J Neurol Sci. 2009;286:18–23. https://doi.org/10.1016/j.jns.2009.08.045.
Matiello M, Kim HJ, Kim W, Brum DG, Barriera AA, Kingsbury DJ, et al. Familial neuromyelitis optica. Neurology. 2010;75:310–5. https://doi.org/10.1212/WNL.0b013e3181ea9f15.
Ghezzi A, Bergamaschi R, Martinelli V, Trojano M, Tola MR, Merelli E, et al. Clinical characteristics, course and prognosis of relapsing Devic’s Neuromyelitis Optica. J Neurol. 2004;251:47–52. https://doi.org/10.1007/s00415-004-0271-0.
Drori T, Chapman J. Diagnosis and classification of neuromyelitis optica (Devic’s syndrome). Autoimmun Rev. 2014;13:531–3. https://doi.org/10.1016/j.autrev.2014.01.034.
Merle H, Olindo S, Bonnan M, Donnino A, Richer R, Smadja D, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology. 2007;114:810–5. https://doi.org/10.1016/j.ophtha.2006.06.060.
Cabre P, González-Quevedo A, Bonnan M, Saiz A, Olindo S, Graus F, et al. Relapsing neuromyelitis optica: long term history and clinical predictors of death. J Neurol Neurosurg Psychiatry. 2009;80:1162–4. https://doi.org/10.1136/jnnp.2007.143529.
•• Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll WM, Chitnis T. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–9. https://doi.org/10.1212/WNL.0000000000001729. An important consensus statement summarizingaccumulated clinical and research findings supporting the diagnosis of NMOSD.This is valuable to the clinician in allowing for early diagnosis.
Sherman E, Han MH. Acute and chronic management of neuromyelitis optica spectrum disorder. Curr Treat Options Neurol. 2015;17:48. https://doi.org/10.1007/s11940-015-0378-x.
• Bonnan M, Valentino R, Debeugny S, Merle H, Ferge JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89:346–51. https://doi.org/10.1136/jnnp-2017-316286. Emphasizes the importance of maintaining an early clinical suspicion of NMO in the appropriate clinical setting, and ensuring that early plasma exchange is provided to increase the likelihood of clinical recovery.
Weinshenker BG, O’Brien PC, Patterson TM, Noseworthy JH, Lucchinetti CF, Dowdick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46(6):878–86.
•• Pittock SJ, Berthele A, Fujihara K, Jin Kim H, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:614–25. https://doi.org/10.1056/NEJMoa1900866. A clinical trial summarizing a newly-approved therapy for NMOSD.
•• Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock S, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394:1352–63. https://doi.org/10.1016/S0140-6736(19)31817-3. A clinical trial summarizing a newly-approved therapy for NMOSD.
•• Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:2114–24. https://doi.org/10.1056/NEJMoa1901747. A clinical trial summarizing a newly-approved therapy for NMOSD.
•• Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19:402. https://doi.org/10.1016/S1474-4422(20)30078-8. A clinical trial summarizing a newly-approved therapy for NMOSD.
Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264:2003–9. https://doi.org/10.1007/s00415-017-8590-0.
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19:298–306. https://doi.org/10.1016/S1474-4422(20)30066-1.
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020;19:391. https://doi.org/10.1016/S1474-4422(20)30070-3.
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stogbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72:756–63. https://doi.org/10.1001/jamaneurol.2015.0533.
Papeix C, Vidal JS, de Seze J, Pierrot-Deseilligny C, Tourbah A, Stankoff B, et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler. 2007;13:256–9. https://doi.org/10.1177/1352458506070732.
Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H, et al. IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75:1423–7. https://doi.org/10.1212/WNL.0b013e3181f8832e.
Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans R, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79:1065–6. https://doi.org/10.1212/WNL.0b013e31826845fe.
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113–5. https://doi.org/10.1177/1352458511431973.
Nechema Y, Moreh E, Weingarden H, Bloch A, Givon U, Vaknin-Dembinski A, et al. Effectiveness of multi-disciplinary rehabilitation for patients with neuromyelitis optica. J Spinal Cord Med. 2016;39(3):311–6. https://doi.org/10.1179/2045772315Y.0000000060.
Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, Olsen CS, et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology. 2016;86:245–52. https://doi.org/10.1212/WNL.0000000000002283.
Fragoso YD, Sousa NAC, Saad T, Alves-Leon SV, Pimentel MLV, Goncalves MVM, et al. Clinical characteristics of patients with neuromyelitis optica spectrum disorders with early onset. J Child Neurol. 2019;34:487–90. https://doi.org/10.1177/0883073819842421.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurologic Ophthalmology and Otology
Rights and permissions
About this article

Cite this article
Glisson, C.C. Neuromyelitis Optica Spectrum Disorders (NMOSD). Curr Treat Options Neurol 24, 241–251 (2022). https://doi.org/10.1007/s11940-022-00709-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-022-00709-4