
Abstract
Purpose of Review
Immune checkpoint inhibition for treatment of metastatic cancers is recognized to cause diverse immune-mediated neurological syndromes. This review will discuss current knowledge about the frequency and varied presentation of these syndromes affecting the peripheral nervous system as well as detail important diagnostic and management considerations.
Recent Findings
Immune-related adverse events affect the peripheral nervous system more often than the central nervous system, and rates are likely underestimated. Most data regarding neurological immune–related adverse events are retrospective, and prospective studies are needed. These immune-mediated peripheral nervous system syndromes can be severe and contribute to mortality. Discontinuation of ICI therapy combined with aggressive medical management can improve outcomes. Data to inform evidence-based treatment approaches, particularly in moderate to severe events, are needed.
Summary
It is important to recognize the association between immune-mediated peripheral nervous system syndromes and immune checkpoint therapy. These syndromes can be phenotypically diverse, but conditions such as acquired demyelinating neuropathy, myositis, and myasthenia gravis predominate. It is important for neurologists to recognize and promptly diagnose these conditions and manage these patients in a multidisciplinary setting to improve outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The beginnings of harnessing a patient’s immune system to fight against cancer date back to the 1890s, when a young American surgeon named William Coley investigated the effect of streptococcal infection on sarcoma (reviewed in [1]). Coley had read published reports describing the regression of inoperable sarcoma tumor as well as other tumors in patients with erysipelas and other infections and subsequently experimented with injecting first streptococci and later heat-inactivated streptococcal and Serratia species (collectively known as Coley’s toxins) for the treatment of inoperable sarcoma. Although he described significant clinical responses, interest in his work faded given inconsistent reporting methodology, poor follow up, and concern regarding reproducibility of his reports combined with the advent of radiation and chemotherapy.
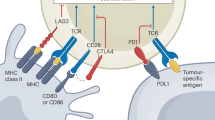
Immune checkpoint inhibition has revolutionized the treatment of cancer since the FDA approval of the first immunotherapy drug ipilimumab for the treatment of metastatic melanoma in 2011. Since that time there has been dramatic development and expanding clinical use of monoclonal antibodies, which enhance the function of anti-tumor T lymphocytes known as immune checkpoint inhibitors (ICIs). ICIs have demonstrated significant impact on morbidity and mortality in various metastatic tumors, though response rates are typically less than 40% [2]. The remarkable impact of these therapies in cancer care was reflected in James Allson and Tasuko Honjo being recognized with the Nobel Prize in Physiology and Medicine in 2018 for their foundational work leading to the development of these novel drugs. At the current time there exist three classes of immunotherapy drugs currently on market: programmed cell death-1 (PD-1) inhibitors (including nivolumab, pembrolizumab, and cemiplimab), programmed cell death ligand-1 (PD-L1) inhibitors (including atezolizumab, avelumab, and durvalumab), and the cytotoxic T lymphocyte associated antigen 4 (CTLA-4) inhibitor ipilimumab. These drugs may be given as monotherapy or in combination. The 7 commercially available ICIs in the USA as of 2020 and current indications for these drugs are listed in Table 1.
In general, ICIs are monoclonal antibodies, which enhance endogenous anti-cancer activity by promoting anti-tumor T cell activation and maintenance of anti-tumor T cell effector function, enabling T cells to recognize and attack tumor cells. Blocking down-regulation of the immune response through treatment with ICIs can result in manifestations of autoimmunity affecting multiple organ systems, a consequence of autoreactive T as well as B cell expansion. These emergent autoimmune conditions following ICI therapy are known as immune-related adverse events (ir-AEs).
A majority of ICI-treated patients experience immune-related adverse events (ir-AEs), reportedly more frequent associated with anti-CTLA-4 Ab treatment (90%) compared with either anti-PD-1 or anti-PD-L1 therapies (70%) [3,4,5]. One proposed explanation for these observed patterns, notably the higher rates of ir-AEs with CTLA-4 inhibitors, is that CTLA-4 is involved in early T cell activation and is expressed predominately in lymphoid tissues, whereas PD-1 and its ligand, PD-L1, are involved in later stages of T cell regulation with higher expression levels in peripheral tissues [6]. Perhaps not unexpected, combination therapy is associated with more frequent ir-AEs than is monotherapy. Dermatologic and gastrointestinal adverse events are particularly common, but not all may be immune-related. The incidence of rash and pruritis has been estimated in up to 40–60%, hepatitis in 5–10%, and diarrhea or colitis in 2–7% of treated patients [7]. Other common ir-AEs include thyroiditis and pneumonitis, which have been more common with anti-PD1 therapies. Interestingly, despite the associated morbidity of these immune-mediated adverse events, the presence of ir-AEs has been repeatedly demonstrated to be associated with improved outcomes in regard to control of the underlying malignancy [8,9,10,11,12].
Multiple neurological ir-AEs affecting either the central or peripheral nervous system have been reported, and sometimes overlapping neurological toxicities can co-exist. While neurological ir-AEs comprise a relative minority of overall ICI-related toxicities, these syndromes can be severe and associated with mortality. They may evolve rapidly and require expeditious workup to determine if the condition is related to cancer progression, radiation toxicity, other chemotherapies, infection, or other metabolic or nutritional derangements. Neurologists need to recognize that these syndromes typically present in an acute to subacute manner and that there is not one specific presentation but that many neuroimmunologic syndromes can result from ICI therapy. Neuromuscular immune-related toxicities appear to be more frequent than those affecting the peripheral nervous system [13,14,15]. As the clinical use of ICIs has expanded with an increasing array of indications for use, the frequency of neurological ir-AEs is expected to increase. It is not clear at this time that patients with pre-existing autoimmune diseases preclude treatment with ICIs if they have a clinical indication to do so [16]. As such, it is important to recognize and treat these syndromes to reduce morbidity and mortality. It is also important to recognize what syndromes do not preclude ongoing ICI therapy, as in many cases this represents salvage therapy for patients without many treatment options. Most published literature describing phenotypes of various neurological syndromes are limited to retrospective case reports, case series, and systematic reviews. The overall purpose of this review is to describe neuromuscular syndromes associated with checkpoint inhibitor toxicity, describe common clinical phenotypes documented in the literature, and discuss proposed therapeutic approaches.
Neurological immune-related adverse events
Two systematic reviews including clinical trials and case reports estimated that in clinical trials, neurological ir-AEs of at least moderate severity comprise less than 1% of overall ICI-related toxicities [17, 18]. In the study reported by Cuzzubbo et al., the frequency of any neurological ir-AE on ICI monotherapy was estimated to be between 3 and 6%, increasing to 12% on combination therapy [17]. Considering neurological AEs of any grade, frequency of neurological events varied between 0 and 27% between clinical trials, highlighting variability amongst studies [18]. Many low-grade neurological complaints are nonspecific, such as fatigue, headache, myalgia, and paresthesia. Comparison of neurological ir-AE rates amongst clinical trials is difficult as thresholds for reporting and definitions for severity differ across studies. Additionally, classification of specific symptoms (e.g., fatigue, myalgia, ocular, or musculoskeletal) varies from clinical trial to clinical trial. Retrospective single and multi-institution case series report neurological ir-AE rates of between 2 and 3% [13, 15, 19]. Estimates of neurological ir-AEs vary considerably across studies based on variability of case definitions in clinical trials as well as reporting bias in case reports and retrospective case series.
Most published literature describing phenotypes of various neurological syndromes are limited to retrospective case reports, case series, and systematic reviews. Central nervous system ir-AEs include aseptic meningitis, hypophysitis, autoimmune encephalopathy, and multiple sclerosis, amongst others. In the peripheral nervous system acute demyelinating polyneuropathy, painful sensorimotor axonal polyneuropathy, mononeuritis multiplex, small fiber neuropathy, dorsal root ganglionopathy, and myasthenia gravis have been described. Neurological ir-AEs are more commonly seen affecting the peripheral nervous system [13, 14]. Neurological signs and symptoms typically present within 6 weeks of initiating ICI therapy, though onset has been described as soon as after the first dose as well as over a year since the commencement of therapy.
Polyneuropathy
Introductory case
A 73-year-old female with a history of right sinus melanoma status post-extensive surgical resection was found to have liver metastasis. She received one cycle of nivolumab and within days developed neck pain and left arm weakness associated with numbness and burning dysesthetic pain in the hand and arm. After approximately 2 weeks she developed right wrist drop and dysesthetic pain in the right hand. A brain MRI with and without contrast revealed post-surgical changes with the interval development of an enhancing, diffusion restricting soft tissue nodule in the lateral aspect of the right maxillary sinus. Electrodiagnostic studies showed evidence for asymmetric, upper extremity sensorimotor axonal mononeuropathies as seen in mononeuritis multiplex. Laboratory studies revealed negative ANA IFA, double-stranded DNA, and ANCA, and she showed no evidence for hepatitis C infection. She was placed on high-dose oral prednisone with plans for plasma exchange or rituximab dependent on clinical course. Pregabalin was commenced for neuropathic pain. After 2 weeks the patient decided to pursue hospice care.
Neuropathies
It has been estimated that neuropathies constitute approximately one-third of all ICI-related neuromuscular ir-AEs [20]. Overall incidence of neuropathy has been estimated at 0.7%, with variability amongst treatment regiments: anti-PD1 or PD-L1 0.3%, anti-CLTA4 1.1%, and combination therapy 1.6% [21•]. Immune-mediated demyelinating neuropathies, such as Guillain–Barre syndrome (GBS), have been reported to comprise the most common neuropathy phenotype particularly in moderate-severe cases [22••]. Acute-onset chronic inflammatory demyelinating polyneuropathy (CIDP) has also been described but is uncommon [20, 23]. Also common is an acute painful axonal polyneuropathy as well as varied cranial neuropathies (including oculomotor palsies, trigeminal neuropathy, trigeminal neuralgia, facial neuropathy, and sensorineural hearing loss), which are typically associated with aseptic meningitis [21•, 24••]. Less common reported neuropathic syndromes include mononeuritis multiplex, brachial plexopathy, and sensory neuronopathy [25, 26].
The median time from initiation of ICI therapy to neuropathy onset is reported to vary between 4 and 10 weeks [15, 21•, 22••]. Ir-AEs affecting other organ systems, such as enterocolitis and pneumonitis, may be noted in over half of patients presenting with neuropathy, noting that neuropathy is the initial irAE [21•]. For the purposes of discussion, the more common neuropathy phenotypes, GBS and painful axonal polyneuropathy, will be considered though peripheral nervous system vasculitis, as described in the case vignette is a rare but serious neurological ir-AE that will also be discussed. We believe that electrodiagnostic evaluation is important in evaluation of patients with large fiber neuropathy syndromes to define the localization and underlying physiology of the disorder (e.g., axonal or demyelinating). It should be noted that the timing of the study is important, as studies performed early after symptom onset (e.g., within 3 weeks) may not have electrodiagnostic findings that have fully evolved to allow for precise localization and determination of severity in the setting of axonal loss.
In GBS the most common phenotype is an acquired inflammatory demyelinating polyneuropathy, though axonal variants as well as Miller–Fisher syndrome (trio of opthalmoparesis, ataxia, and areflexia associated with GQ1b Ab) have been reported. Spinal fluid may show typical features of albuminocytologic disassociation (elevated total protein with normal cell count) but it is not uncommon (reported in up to half of the affected patients) to have a lymphocytic pleocytosis with up to 100 cells [20, 22••, 27]. We consider it important to include cytology and remember to obtain a serum blood glucose at the time of the lumbar puncture to evaluate for possible leptomeningeal spread of metastatic disease. GBS is not associated with hypoglycorrhachia, and low CSF glucose should prompt a reconsideration of a GBS diagnosis. Cases of polyradiculopathy have been reported, and at this point it is unclear if these represent a manifestation of GBS or a radiculitis associated with leptomeningeal inflammation as is the case with cranial neuropathies [15, 24••]. In a recent review of published case series and case reports, MRI has shown root enhancement in approximately a quarter of ir-AE GBS patients [20]. There exists limited data regarding neuropathology, but segmental demyelination and perivascular inflammation have been reported [20]. Although treatment considerations are to be discussed later, it is important to recognize that management of these patients includes treatment with high-dose corticosteroids as is recommended in treatment of ICI ir-AEs, which contrasts with treatment recommendations for idiopathic GBS wherein steroids are not recommended. [23, 28].
The most prominent feature of ICI-related axonal polyneuropathy (which may be sensory or sensorimotor) is that of neuropathic pain. A retrospective review has suggested that all patients with this neuropathy phenotype have mild disease severity [29]. Cessation of ICI therapy and analgesic therapy has been reported to lead to satisfactory improvement in almost all affected patients [15].
Peripheral nervous system vasculitis is rare; however, there are no clear estimates as to the frequency of vasculitis amongst ICI-treated patients as most information regarding this clinical entity is relegated to case reports. A systematic review of the literature identified 53 reported cases of vasculitis, with data sufficient to confirm the diagnosis in 20 cases [25]. Large vessel vasculitis (e.g., giant cell arteritis, aortitis) and vasculitis affecting either the CNS or PNS were most common. Of the described cases, isolated peripheral nervous system vasculitis was observed in 15%. Conditions to be considered include polyarteritis nodosa from hepatitis B infection, hepatitis C-associated cryoglobulinemic vasculitis, and HIV as well as other infectious vasculitis. The presence of ANCA, MPO, or PR3 may indicate ANCA-associated vasculitis, and patients should be followed for development of systemic manifestations.
Neuromuscular junction disorders
Introductory case
A 66-year-old man with stage IV lung cancer on immunotherapy with pembrolizumab presented to an outside hospital with 1 week of worsening ptosis, diplopia, dysphagia and dyspnea. Upon admission he was found to have significant respiratory compromise and was intubated. Workup revealed positive AchR binding antibodies (2.97 nmol/L, normal < 0.02) and normal CK. He received two courses of IVIg and steroids at the OSH without any improvement and needing now a tracheostomy. He was transferred to Emory University Hospital. On exam he had marked ptosis, ophthalmoparesis, and proximal weakness. Here he received 5 courses of plasma exchange and was started on Rituximab. He had some mild improvement in limb strength and started to be able to be weaned of the ventilator. He was discharged to acute rehabilitation and Rituximab infusions continued. Upon follow up, 2 weeks from discharge, ptosis and diplopia had resolved; patient was now using a trach collar and was starting to walk with the use of a walker.
Myasthenia gravis
Myasthenia gravis is one of the most commonly reported neuromuscular disorders associated with ICI therapy. Incidence is not fully known and reports of incidence of myasthenia gravis range from 0.12 to 0.2% [14, 30•, 31•].
ICI-associated myasthenia gravis can present as new onset myasthenia gravis (ocular or generalized), myasthenia gravis crisis, worsening of known previously diagnosed myasthenia, or an overlap syndrome of myasthenia gravis associated with myositis and/or myocarditis. Approximately two third of cases are de novo myasthenia gravis; the remainder had the preexisting condition [32]. While there is not clear evidence that pre-existing autoimmune conditions preclude a treatment trail with an ICI, in the setting of myasthenia we consider prudent to have the myasthenia under good control prior to the treatment trial, as some patients have limited treatment options for their cancer outside of ICI therapy. Both acetylcholine receptor antibody positive and seronegative cases have been reported; to date, there are no reports of muscle-specific kinsase (MusK) antibody myasthenia gravis [33]. There are also no reports on Lambert–Eaton myasthenic syndrome.
Most cases of myasthenia gravis published in the literature are with the use of ipilimumab, nivolumab, or pembrolizumab, given either as monotherapy or combination therapy ICIs are administered as infusions every 2 to 4 weeks, but this varies depending on toxicity and indication. The mean time from treatment to onset of symptoms is 5.8 weeks (± 4 weeks) [33], with a range reported from 2 to 15 weeks [32]. The number of doses of ICIs is, in average, 2.4 [32].
We have seen in our clinic and in our university hospital de novo myasthenia (ocular, generalized, crisis) and exacerbation of preexisting myasthenia. Most have had a favorable outcome, but we have had at least two fatal cases, one of which suffered severe coexistent cardiomyopathy from myocarditis.
Introductory case
A 73-year-old male with a history of renal cell carcinoma treated with nivolumab presented with fatigue, diplopia, weakness, and myalgia within 10 days after starting nivolumab. Brain MRI demonstrated a remote lacunar infarct in the left frontal lobe but otherwise was nonrevealing. He received intravenous methylprednisolone for 2 days and was discharged on high-dose oral prednisone for autoimmune hepatitis. Acetylcholine receptor antibodies were sent. Three days after discharge, he was found to have persistently elevated transaminases and was started on mycophenolate for autoimmune hepatitis. Electrodiagnostic studies with slow repetitive nerve stimulation did not show an electrodecrement. Creatine kinase was elevated (5001 unit/L; normal range 30–223), and he was readmitted and started on methylprednisolone in combination with intravenous immunoglobulin (IVIg) due to concern for autoimmune myositis. After he complained of chest pain and dyspnea, troponin found to be elevated at 26.6 mg/mL. Echocardiogram demonstrated 40% ejection fraction with basal inferior and anterior hypokinesis. Cardiac catherization revealed 80% stenosis of the proximal LAD, and the patient received a drug eluding stent given concern for cardiac ischemia. IVIg was discontinued after one dose given risk of thrombosis. His dyspnea progressed, and the patient required intubation and progressive weakness, while reflexes and sensation remained intact. This was presumed to be secondary to myasthenic syndrome, and he began plasma exchange, which was not tolerated due to hypotension.
Although he demonstrated some short-term clinical improvement with regard to weakness, the course was complicated by atrioventricular disassociation with bradycardia and subsequent hemodynamic compromise, requiring an ICD and pacemaker. Acetylcholine antibodies resulted consistent with autoimmune myasthenia gravis (acetylcholine receptor binding antibody 16.1 nmol/L (normal < 0.2) and acetylcholine receptor modulating antibody 94%). Striational antibody testing was elevated at 1:61,440. A myositis antibody panel and HMG CoA reductase antibody were negative. He was treated with rituximab; however, a complicated hospital course ensued, and he ultimately succumbed to multiorgan failure almost 3 months from his initial presentation; overall, his clinical course and laboratory markers suggested an ICI-related myasthenia-fulminant myositis syndrome.
Inflammatory myopathy
Myalgia is a commonly identified symptom in individuals receiving checkpoint inhibitors, occurring with an estimated prevalence of 2–21% [34]. However, de novo inflammatory myopathies are far less frequent. A retrospective study of 1293 patients who were treated with a checkpoint inhibitor identified 10 patients who developed myositis (prevalence of 0.8%) [35]. Meanwhile, other studies including patients who only received PD-1 inhibitor therapies identified muscle disorders in 0.58 to 0.76% of patients [13, 31•]. The most common inflammatory myopathies triggered by checkpoint inhibitors include necrotizing autoimmune myositis, dermatomyositis, polymyositis, and granulomatous myositis [36•, 37].
Symptoms of myositis range from myalgias, weakness typically in limb-girdle pattern, axial weakness (most predominantly at cervical level with neck extensor weakness), to ptosis, and oculomotor weakness with diplopia [32, 38••]. As described in the neuromuscular junction disorders section, and as our case demonstrates, there is common overlap with myasthenia gravis. Suzuki et al. found in a series of 12 patients with myasthenia gravis that 4 had concomitant myositis. Symptoms of myositis tend to occur at the same time as myasthenic symptoms [30•]. Conversely, Dubey et al. identified 28 patients with grade III or IV neurologic adverse events. Thirty-six percent (10 patients) developed myositis. Of those 10, half (5 patients) had a myositis-myasthenia overlap syndrome [24••]. This finding highlights the importance of considering repetitive nerve stimulation to evaluate for a disorder of neuromuscular transmission even in the setting of elevated CK and consideration of ordering Ach-R Abs in patients with weakness that impairs function as there may be more than one process driving the clinical syndrome.
However, inflammatory myositis associated with checkpoint inhibitors appears to have the unique ability to involve ocular muscles, even in the absence of concomitant myasthenia gravis [31•]. This feature of ocular involvement is otherwise not commonly seen in inflammatory and necrotizing myopathies occurring in patients without checkpoint inhibitor exposure and may present a diagnostic conundrum in attempting to distinguish from myasthenia. Additionally, the heart is often not spared, with one review identifying myocarditis in 16% out of 180 patients with myositis while another study identified myocarditis in 40% (four patients) [35, 39]. We consider checking troponin, especially at baseline and afterwards as clinically warranted, as well as a transthoracic echocardiogram in patients presenting with moderate–severe myopathic ir-AEs.
In a retrospective analysis of patients treated with checkpoint inhibitors and who developed myositis (n = 10) in tertiary centers in Paris, France, and Berlin, Germany, from January 2015 to July 2017, Touat et al. identified a median onset of myositis from ICI initiation of 25 days (range 5 to 87 days) [38••]. Similarly, the World Health Organization Individual Case Safety Report pharmacovigilance database identified a median time to onset of symptoms after initiation of treatment of 26 days in 180 patients with myositis [39]. In the Touat et al. study, eight patients developed myositis following the second round of infusion of immunotherapy as monotherapy. In the remaining two patients, both patients that developed myositis after the first infusion were treated with combined immunotherapy (nivolumab and ipilimumab) as opposed to monotherapy. Similarly, a study by Dubey et al. found that combination therapy with anti-PD1/PD-L therapy plus anti-CTLA-4 is associated with a higher risk of neurologic immune–related adverse events than in those receiving anti-PD1/PD-L1 monotherapy [24••].
Laboratory testing tends to reveal elevation in creatine kinase levels with range of 72 to 30,980 U/L and mean value of 6126 [32]. Myositis antibodies tend to be absent, although occasional reports of striational antibodies may be elevated, and cases of positive anti-MDA5 antibody and anti-SRP antibody are identified [40]. In patients with myositis–myasthenia overlap syndromes, anti-acetylcholine receptor and anti-striated muscle antibodies have been reported, including our case presented above [30•]. We feel it is reasonable to monitor CK values especially early in the course, as there may be confounding factors for weakness in patients with malignancy (poor nutrition, asthenia, and overlap syndromes such as myositis-myasthenia). In patients who are clinically weak we routinely perform electrodiagnostic studies to evaluate for irritable myopathy as well as for the presence of myasthenia. Evaluation of respiratory function may be indicated for patients with moderate–severe muscle weakness.
Biopsy of affected muscle typically demonstrates focal endomysial inflammatory infiltrates, which consist primarily of CD8-positive and CD68 positive cells, as well as to a lesser extent CD4-positive cells [30•, 38••]. Macrophages, as identified with CD68 markers, are the most abundant cells identified in intramuscular inflammatory exudate in granulomatous myositis. PD-1 expression identified on tumor-infiltrating macrophages suggests that the pathophysiology of at least a subset of the inflammatory myositis seen in patients receiving checkpoint inhibitors may not be limited to T cells [32, 41]. As muscle biopsy is unlikely to change immediate course of management (particularly in a patient with weakness, elevated CK, and electrodiagnostic studies showing irritable myopathy), we recommend prompt initiation of treatment with consideration of biopsy only if patient has progression despite appropriate therapy or severe, refractory disease.
Current treatment
Treatment of emergent neuromuscular complications associated with ICI therapy depends on the severity of the condition, inherent risks of treatment, and comorbid conditions. Management includes the choice of therapy for the neuromuscular disorder itself and the decision on whether to withhold ICIs or not. There is no standardized therapy and most neurologists tailor their treatment approach based on the severity of the complications with attention the condition’s impact on function. Side effects are generally classified as grade 1 (asymptomatic or mild), grade 2 (moderate, affecting ADLs), and grades 3–4 (severe, life-threatening) [42]. Practice recommendations and guidelines for management of ir-AEs exist that we have found helpful for guiding the decision-making process [43,44,45,46]. Our general approach to management of more common neuromuscular ir-AE management is detailed in Table 2. Myasthenia gravis, inflammatory myopathies, and acquired demyelinating neuropathies are all generally considered serious events, whereas paresthesias and small fiber sensory neuropathies are considered mild events [36•].
For grade 1 neurological ir-AEs no treatment is recommended outside of symptomatic therapies (e.g., analgesics for neuropathic pain, pyridostigmine for myasthenia), and ICI treatment may be continued with careful monitoring. For grade 2 events the ICIs should be held and steroids are recommended as first line therapy [42]. When symptoms improve to grade 1 or less, then considering resumption of ICI treatment can be entertained, noting that the treating oncologist would make the final decision. For grade 3 events most guidelines recommend discontinuation of ICI drugs and first line therapy involves the use of rapid action treatments such as corticosteroids (either 1 g/d IV solumedrol for 3–5 days or oral prednisone 1 mg/kg), intravenous immunoglobulin (IVIG) 2 g/kg, or plasma exchange (PLEX) 5–7 exchanges [45]. Pyridostigmine should be initiated at 60 mg three times daily for myasthenia and titrated as needed with hyoscyamine for cholinergic side effects (abdominal discomfort, diarrhea, cramping). It is important to note that steroids are indicated for the treatment ICI-related GBS, frequently in combination with either IVIG or PLEX. In moderate–severe myasthenia gravis it is important to realize that high-dose steroids can result in paradoxical clinical worsening, which usually occurs about 7–10 days after initiation of treatment, and if high-dose steroids are given we typically also treat with either IVIG or PLEX to safeguard against steroid-associated worsening. In the outpatient setting, for myasthenia oral steroids are started at a dose of 10–20 mg and titrated by 10 mg weekly to 40–60 mg daily. In our experience, PLEX is favored over IVIG in myasthenia gravis exacerbations or crisis as well as for the treatment of more severe myopathies and neuropathies without significant time constraints given that these more severe cases are often hospitalized for treatment. We have used IVIG in grade 2 myopathies or large fiber acquired demyelinating neuropathies that are more chronic in progression and that remain ambulatory. Specific management will need to be individualized depending on the severity and response to therapy and escalated or tapered as deemed appropriate, and careful evaluation of respiratory forces is important for moderate-severe GBS, myasthenia, and myopathy. In the neuropathy case we presented, steroids were given with the plan to use more aggressive therapy depending on her response; the myasthenia/myositis overlap case did not respond to IVIG so PLEX and rituximab were given; unfortunately, the myositis overlap case failed multiple treatment options. A multidisciplinary approach with a neurologist with frequent neurological follow-up should guide therapeutic decisions.
Initial immunomodulatory treatments may need to be followed by long-term immunosuppression, which will also be dependent on the severity of the condition and response to first line therapies. Long-term immunosuppressants include azathioprine, mycophenolate mofetil, rituximab, and methotrexate. Reports of the long term natural history and response to treatment for these neuromuscular complications are lacking, and our experience is to choose the long-term therapy as we would for the same neuromuscular condition not caused by ICIs [36•]. The National Comprehensive Cancer Network guidelines suggest the use of rituximab in severe myasthenia gravis [46].
The fate of ICI treatment depends on the severity of the neurologic complication and the status of underlying malignancy. In cases of mild events it may be possible to manage symptomatically and continue the immune therapy uninterrupted or after holding one or two doses to allow for stabilization of the symptoms. However, given the potential morbidity associated with severe neurologic disease and subsequent exacerbation following retrial of ICIs, frequently, the ICI is stopped and other treatment options if available are sought. This requires a multidisciplinary discussion and collaboration between the neurologist, treating oncologist and the patient.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39. https://doi.org/10.1016/j.intimp.2018.06.001.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. https://doi.org/10.1056/NEJMoa1003466.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. https://doi.org/10.1056/NEJMoa1200690.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. https://doi.org/10.1056/NEJMoa1200694.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106. https://doi.org/10.1097/COC.0000000000000239.
Marin-Acevedo JA, Chirila RM, Dronca RS. Immune checkpoint inhibitor toxicities. Mayo Clin Proc. 2019;94(7):1321–9. https://doi.org/10.1016/j.mayocp.2019.03.012.
Eggermont AMM, Kicinski M, Blank CU, Mandala M, Long GV, Atkinson V, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2020;6:519. https://doi.org/10.1001/jamaoncol.2019.5570.
Das S, Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer. 2019;7(1):306. https://doi.org/10.1186/s40425-019-0805-8.
Masuda K, Shoji H, Nagashima K, Yamamoto S, Ishikawa M, Imazeki H, et al. Correlation between immune-related adverse events and prognosis in patients with gastric cancer treated with nivolumab. BMC Cancer. 2019;19(1):974. https://doi.org/10.1186/s12885-019-6150-y.
Grangeon M, Tomasini P, Chaleat S, Jeanson A, Souquet-Bressand M, Khobta N, et al. Association between immune-related adverse events and efficacy of immune checkpoint inhibitors in non-small-cell lung Cancer. Clin Lung Cancer. 2019;20(3):201–7. https://doi.org/10.1016/j.cllc.2018.10.002.
Ricciuti B, Genova C, De Giglio A, Bassanelli M, Dal Bello MG, Metro G, et al. Impact of immune-related adverse events on survival in patients with advanced non-small cell lung cancer treated with nivolumab: long-term outcomes from a multi-institutional analysis. J Cancer Res Clin Oncol. 2019;145(2):479–85. https://doi.org/10.1007/s00432-018-2805-3.
Kao JC, Liao B, Markovic SN, Klein CJ, Naddaf E, Staff NP, et al. Neurological complications associated with anti-programmed death 1 (PD-1) antibodies. JAMA Neurol. 2017;74(10):1216–22. https://doi.org/10.1001/jamaneurol.2017.1912.
Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:210–25. https://doi.org/10.1016/j.ejca.2016.02.024.
Bruna J, Argyriou AA, Anastopoulou GG, Alemany M, Nadal E, Kalofonou F, et al. Incidence and characteristics of neurotoxicity in immune checkpoint inhibitors with focus on neuromuscular events: experience beyond the clinical trials. J Peripher Nerv Syst. 2020;25:171–7. https://doi.org/10.1111/jns.12371.
Tison A, Quere G, Misery L, Funck-Brentano E, Danlos FX, Routier E, et al. Safety and efficacy of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune disease: a Nationwide, Multicenter Cohort Study. Arthritis Rheum. 2019;71(12):2100–11. https://doi.org/10.1002/art.41068.
Cuzzubbo S, Javeri F, Tissier M, Roumi A, Barlog C, Doridam J, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1–8. https://doi.org/10.1016/j.ejca.2016.12.001.
Mohn N, Beutel G, Gutzmer R, Ivanyi P, Satzger I, Skripuletz T. Neurological immune related adverse events associated with nivolumab, ipilimumab, and pembrolizumab therapy-review of the literature and future outlook. J Clin Med. 2019;8(11). https://doi.org/10.3390/jcm8111777.
Spain L, Walls G, Julve M, O'Meara K, Schmid T, Kalaitzaki E, et al. Neurotoxicity from immune-checkpoint inhibition in the treatment of melanoma: a single centre experience and review of the literature. Ann Oncol. 2017;28(2):377–85. https://doi.org/10.1093/annonc/mdw558.
Puwanant A, Isfort M, Lacomis D, Zivkovic SA. Clinical spectrum of neuromuscular complications after immune checkpoint inhibition. Neuromuscul Disord. 2019;29(2):127–33. https://doi.org/10.1016/j.nmd.2018.11.012.
• Dubey D, David WS, Amato AA, Reynolds KL, Clement NF, Chute DF, et al. Varied phenotypes and management of immune checkpoint inhibitor-associated neuropathies. Neurology. 2019;93(11):e1093–e103. https://doi.org/10.1212/WNL.0000000000008091. This retrospective case series describes diverse phenotypes of peripheral nervous system pathology derived from two tertiary institutions.
•• Chen X, Haggiagi A, Tzatha E, DeAngelis LM, Santomasso B. Electrophysiological findings in immune checkpoint inhibitor-related peripheral neuropathy. Clin Neurophysiol. 2019;130(8):1440–5. https://doi.org/10.1016/j.clinph.2019.03.035. A retrospective single-institution case series which is the first to describe detailed electrodiagnostic features of ICI-related peripheral neuropathies.
Supakornnumporn S, Katirji B. Guillain-Barre syndrome triggered by immune checkpoint inhibitors: a case report and literature review. J Clin Neuromuscul Dis. 2017;19(2):80–3. https://doi.org/10.1097/CND.0000000000000193.
•• Dubey D, David WS, Reynolds KL, Chute DF, Clement NF, Cohen JV, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol. 2020;87(5):659–69. https://doi.org/10.1002/ana.25708. This study describes all severe neurologic adverse events associated with immune checkpoint inhibitors experienced at a tertiary care institution, including neuromuscular disease as well as overlap syndromes. Moreover, the article includes an in-depth discussion on management.
Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin Rheumatol. 2018;37(9):2579–84. https://doi.org/10.1007/s10067-018-4177-0.
Alhammad RM, Dronca RS, Kottschade LA, Turner HJ, Staff NP, Mauermann ML, et al. Brachial plexus neuritis associated with anti-programmed cell death-1 antibodies: report of 2 cases. Mayo Clin Proc Innov Qual Outcomes. 2017;1(2):192–7. https://doi.org/10.1016/j.mayocpiqo.2017.07.004.
Gu Y, Menzies AM, Long GV, Fernando SL, Herkes G. Immune mediated neuropathy following checkpoint immunotherapy. J Clin Neurosci. 2017;45:14–7. https://doi.org/10.1016/j.jocn.2017.07.014.
Hughes RA, Wijdicks EF, Barohn R, Benson E, Cornblath DR, Hahn AF, et al. Practice parameter: immunotherapy for Guillain-Barre syndrome: report of the quality standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61(6):736–40. https://doi.org/10.1212/wnl.61.6.736.
Chen JH, Lee KY, Hu CJ, Chung CC. Coexisting myasthenia gravis, myositis, and polyneuropathy induced by ipilimumab and nivolumab in a patient with non-small-cell lung cancer: a case report and literature review. Medicine (Baltimore). 2017;96(50):e9262. https://doi.org/10.1097/MD.0000000000009262.
• Suzuki S, Ishikawa N, Konoeda F, Seki N, Fukushima S, Takahashi K, et al. Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology. 2017;89(11):1127–34. https://doi.org/10.1212/WNL.0000000000004359. Describes a series of patients with PD-1 associated myasthenia gravis, detailing the myasthenia – myositis – myocarditis overlap syndrome.
• Liewluck T, Kao JC, Mauermann ML. PD-1 inhibitor-associated myopathies: emerging immune-mediated myopathies. J Immunother. 2018;41(4):208–11. https://doi.org/10.1097/CJI.0000000000000196. A case series of PD-1 associated myopathies which describes the unique features of ocular and bulbar involvement as can be seen in ICI-associated myositis.
Kao JC, Brickshawana A, Liewluck T. Neuromuscular complications of programmed cell death-1 (PD-1) inhibitors. Curr Neurol Neurosci Rep. 2018;18(10):63. https://doi.org/10.1007/s11910-018-0878-7.
Kolb NA, Trevino CR, Waheed W, Sobhani F, Landry KK, Thomas AA, et al. Neuromuscular complications of immune checkpoint inhibitor therapy. Muscle Nerve. 2018;58:10–22. https://doi.org/10.1002/mus.26070.
Abdel-Wahab N, Suarez-Almazor ME. Frequency and distribution of various rheumatic disorders associated with checkpoint inhibitor therapy. Rheumatology (Oxford). 2019;58(Suppl 7):vii40–vii8. https://doi.org/10.1093/rheumatology/kez297.
Richter MD, Crowson C, Kottschade LA, Finnes HD, Markovic SN, Thanarajasingam U. Rheumatic syndromes associated with immune checkpoint inhibitors: a single-center cohort of sixty-one patients. Arthritis Rheum. 2019;71(3):468–75. https://doi.org/10.1002/art.40745.
• Dalakas MC. Neurological complications of immune checkpoint inhibitors: what happens when you 'take the brakes off' the immune system. Ther Adv Neurol Disord. 2018;11:1756286418799864. https://doi.org/10.1177/1756286418799864. A succinct review article for neurologists describing ICIs, their mechanism of action, proposed reason for autoimmunity, and summarizing all described neurological complications to date of publication.
Uchio NTK, Ikenaga C, Unuma A, Kadoya M, Kubota A, et al. Granulomatous myositis induced by anti-PD-1 monoclonal antibodies. Neurol Neuroimmunol Neuroinflamm. 2018;5(4).
•• Touat M, Maisonobe T, Knauss S, Ben Hadj Salem O, Hervier B, Aure K, et al. Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology. 2018;91(10):e985–e94. https://doi.org/10.1212/WNL.0000000000006124. Excellent article that investigates features of patients who develop myositis, including weakness patterns but also histopathology findings.
Anquetil C, Salem JE, Lebrun-Vignes B, Johnson DB, Mammen AL, Stenzel W, et al. Immune checkpoint inhibitor-associated myositis: expanding the spectrum of cardiac complications of the immunotherapy revolution. Circulation. 2018;138(7):743–5. https://doi.org/10.1161/CIRCULATIONAHA.118.035898.
Diamantopoulos PT, Tsatsou K, Benopoulou O, Anastasopoulou A, Gogas H. Inflammatory myopathy and axonal neuropathy in a patient with melanoma following pembrolizumab treatment. J Immunother. 2017;40(6):221–3. https://doi.org/10.1097/CJI.0000000000000172.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9. https://doi.org/10.1038/nature22396.
Hottinger AF. Neurologic complications of immune checkpoint inhibitors. Curr Opin Neurol. 2016;29(6):806–12. https://doi.org/10.1097/WCO.0000000000000391.
Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;36(17):1714–68. https://doi.org/10.1200/JCO.2017.77.6385.
Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, Dadu R, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5(1):95. https://doi.org/10.1186/s40425-017-0300-z.
Anderson D, Beecher G, Nathoo N, Smylie M, McCombe JA, Walker J, et al. Proposed diagnostic and treatment paradigm for high-grade neurological complications of immune checkpoint inhibitors. Neurooncol Pract. 2019;6(5):340–5. https://doi.org/10.1093/nop/npy039.
Thomson JA SB, Brahmer J, Andrews S, Armand P et al. Managment of immunotherapy-related toxicicites. 2019. https://www.nccn.org/professionals/physician_gls/default.aspx#immunotherapy. Accessed March 6 2020.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuromuscular Disorders
Rights and permissions
About this article

Cite this article
Garcia-Santibanez, R., Khoury, M. & Harrison, T.B. Immune-Related Neuromuscular Complications of Checkpoint Inhibitors. Curr Treat Options Neurol 22, 27 (2020). https://doi.org/10.1007/s11940-020-00635-3
Published:
DOI: https://doi.org/10.1007/s11940-020-00635-3