
Abstract
Purpose of review
The purpose of this review article is to discuss the pathogenesis of acute and chronic immune-mediated neuropathies along with the recent advances in their treatment.
Recent findings
Since the first description of Guillain-Barre syndrome (GBS) more than a century ago, there have been numerous forms of immune-mediated neuropathies described expanding the spectrum. Understanding the role of the immune system in the pathogenesis of immune-mediated neuropathies has been an advancement towards the diagnosis and treatment.
Summary
It is postulated that immune-mediated neuropathies are a group of diseases resulting from autoimmunity towards multiple components of peripheral nervous system. These have a wide range of pathologic mechanisms, defined clinical presentations, electro-diagnostic and laboratory findings which help in diagnosis and management. Although immunosuppression is the common modality of treatment for these disorders, uncovering distinct pathogenic mechanisms can allow for targeted immunomodulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune-mediated neuropathies are a group of diseases resulting from autoimmunity towards multiple components of the peripheral nervous system. Patients usually present with sensory deficits, muscle weakness and gait unsteadiness. There seems to be overlap of symptoms among different subtypes. Time of onset of symptoms helps in identifying acute versus chronic immune-mediated neuropathies. Although the pathogenic mechanisms are not clearly understood in many of these diseases, we do know that the damage is a result of direct attack by autoantibodies to proteins, glycoproteins and glycolipids on neuronal cell membranes and myelin. This group of neuropathies comprises 9–18% of all causes of neuropathy [1].
Guillain-Barre syndrome
Landry first described the clinical features of Guillain-Barre syndrome (GBS) in 1859. In his report, he described 5 patients with all features of GBS in “Traité complet des paralysies” (Complete treatise of palsies). In 1916, Guillain, Barre and Strohl described this illness in two French soldiers during World War I, and that is how it derives its name.
Subacute flaccid paralysis is most commonly caused by GBS [2, 3••]. Diagnosis of GBS is based on clinical and electro-diagnostic features and laboratory findings. The classic clinical features include ascending numbness and weakness over days to weeks, hyporeflexia or areflexia, respiratory insufficiency and dysautonomia. Most patients achieve maximum weakness by the 4th week and then plateau. Worsening weakness after 4 weeks is a red flag for other illness, particularly chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Cranial nerve findings include unilateral or bilateral facial weakness in 50% of patients and ophthalmoparesis in 20% [4,5,6]. Cerebrospinal fluid shows albumino-cytological dissociation. Electro-diagnostic studies show prolonged F-wave latency early on and slowing of conduction velocity.
GBS variants include motor or sensory and demyelinating or axonal. The acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is the most common form of GBS. Other variants include acute motor axonal neuropathy (AMAN) and acute motor sensory axonal neuropathy (AMSAN) where the primary target is the axons and the nodes of Ranvier mediated by macrophages and antibodies. AMAN seems to be associated with anti-GM1 and anti-GD1a antibodies. Patients with AMAN and anti-GM1 antibodies usually have preceding infection with C. Jejuni. Miller Fisher syndrome is a variant characterised clinically by the triad of ataxia, ophthalmoplegia and areflexia and anti-GQ1b antibody. The involvement of dorsal roots and ganglionic neurons can present as sensory ataxic GBS. It is considered to be in the spectrum of Miller Fisher syndrome as these patients have IgG antibodies to GQ1b or GD1b ganglioside. Lastly, acute pandysautonomic neuropathy where the target antigen is the sympathetic ganglionic neurons [7••].
Recent data suggests that GBS is an antibody-mediated disorder [2, 3••, 5, 7••]. The mechanism of antibody production is thought to be from molecular mimicry between the surface molecules of he motor axons and lipooligosaccharides of infective organisms [8••]. The anti-GM1 and anti-GD1a antibodies in AMAN are of the IgG1 and IgG3 subclass and are thought to play a pathogenic role by binding to the gangliosides resulting in axonal injury by complement fixation, macrophage migration and membrane attack complex deposition on the axolemma [3••]. See Figs. 1 and 2.

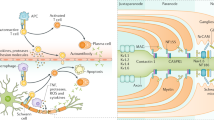
Pathogenesis of immune-mediated neuropathies: Onset of AIDP can be preceded by an infection, due to molecular mimicry (similar structure of bacterial antigen and peripheral nerve component). Antigen presenting cells activate autoreactive T cells which in turn causes conversion of B cell to plasma cell which release autoantibodies; and secretion of cytokines that degrade basement membrane of the blood vessels and disrupt the blood-nerve barrier. This allows the transfer of inflammatory cells and pro-inflammatory cytokines and complement to the peripheral nerve compartment. Upon entering the nerve, the autoantibodies can bind to the myelin glycoproteins or proteins in the nodal or paranodal region such as contactin-associated protein 1 (CASPR1), contactin 1 or Neurofascin 155 (NF155) and gliomedin. (Adapted by permission from Springer Nature: Springer Publishing Company: Nature Reviews Disease Primers: Kieseier BC, Mathey EK, Sommer C, Hartung H-P. Immune-mediated neuropathies. Nat Rev. Dis Prim. 2018;4:31.).

a Segmental demyelination (100×) and b paranodal demyelination in a teased nerve fibre (1000×) in a patient with CIDP. (a, b—Image courtesy Aziz Shaibani, MD) c Osteolytic lesion on a bone radiograph in a patient with POEMS syndrome.
Treatment
The management of GBS involves three components: general medical care, immunomodulation and rehabilitation.
General medical care
Close monitoring is required for respiratory failure with periodic measurements of respiratory reserve, such as forced vital capacity (FVC), maximal inspiratory pressure (MIP) or (NIF) negative inspiratory force > 25 cm H2O and maximal expiratory pressure (MEP). The 20/30/40 rule for intubation is followed—FVC below 20 ml/kg of body weight, MIP below − 30 cm H2O, MEP below 40 cm H2O [9]. Note that arterial blood gas and respiratory rate are poor predictors of respiratory compromise, as they are not affected until advanced stages of the illness. Close monitoring for autonomic disturbances with telemetry for cardiac arrhythmias, labile blood pressure and variation in body temperature is recommended. Dysphagia, constipation, ileus and urinary retention are the other symptoms to watch for.
In patients who are bed bound, prophylaxis for deep vein thrombosis and decubitus ulcers is essential. Inflammation of the roots and nerves can cause significant pain and should be treated with agents that treat neuropathic pain like gabapentin or low-dose tricyclic antidepressants [10]. Narcotics may be used for a short period. Monitoring in intensive care unit versus floor is to be determined based on the severity of above parameters.
Immunomodulation
Early use of intravenous immunoglobulin (IVIg) and/or plasma exchange (PLEX) have been shown to have equal efficacy in patients with GBS [11,11,12,13,15]. The typical dose based on prior studies of IVIg is 0.5 g/kg/day over 4 days. There is no consensus if this schedule is superior to administering the same dose over 2 to 3 days. There are several commercial preparations available; the choice of preparation depends upon patient factors like diabetes mellitus or renal impairment. PLEX is usually performed on alternate days to avoid complications related to fluid shift. Typically 4–6 exchanges (50 ml/kg) over 8–10 days are done.
IVIg is administered peripherally over 4–6 h and is more readily available. PLEX takes more than a week, needs specialised equipment, a pathologist and a central line; though in some centres it is done peripherally. There is no benefit of combining the 2 treatments [15]. Steroids are not beneficial in GBS [16]. See Table 1.
Eculizumab is a monoclonal antibody that is a terminal complement pathway component C5 inhibitor, which was approved for myasthenia gravis in 2017. It has been studied in conjunction with IVIg in 2 small randomised, double blind, placebo controlled trials for GBS. The ICA-GBS study studied 7 patients; of these 5 patients received IVIg plus eculizumab and 2 received IVIg and placebo. They concluded that eculizumab is well tolerated in patients with acute GBS and can be safely given with IVIg [17]. The JET-GBS study which studied eculizumab (n = 23) vs placebo (n = 11) did not show any significant differences in the primary end point [18]. There is currently insufficient evidence to recommend eculizumab in the treatment of GBS, but larger randomised control trials are required to better assess efficacy and safety.
Rehabilitation
In addition to immune therapy, rehabilitation plays an important role in recovery. Initiation of physical and occupational therapy as soon as patient’s symptoms stabilise clinically is important. Starting with functional assessment of the extent of disability and helping the patient gain independence in activities of daily living followed by gradual introduction and increment of exercises involving resistance is done. It is important to note that these patients should not be pushed to exercise more during a particular session if they are fatigued as this might make the weakness worse and retard the progress [19]. It also helps combat fatigue which is an important problem in GBS. Safety measures should include fall prevention.
Chronic inflammatory demyelinating polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) was first described by Eichhorst in 1890 in a patient with clinical presentation similar to GBS, but with a more chronic and relapsing course. The incidence of CIDP is 1.0 to 8.9 persons per 100,000 [20]. Typical CIDP is characterised by symmetric, proximal and distal, motor and sensory deficits with absent deep tendon reflexes. Symptoms evolve over 8 weeks in a progressive or relapsing manner. A minority may have an acute GBS-like presentation. Absence of respiratory and cranial nerve involvement differentiates CIDP from GBS. Atypical CIDP is less common and includes distal acquired demyelinating symmetric neuropathy (DADS), multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) and pure motor and sensory CIDP [21].
DADS is characterised by slowly evolving distal sensory symptoms with sensory ataxia and only mild distal weakness [22]. Two third of cases are associated with an IgM antibody and are considered as distinct from CIDP. (see section on ‘Demyelinating neuropathies associated with monoclonal gammopathy’). The remaining one third of cases are considered as atypical forms of CIDP which also respond to IVIg and corticosteroid therapies. Distal muscles and upper limbs are predominantly affected. MADSAM also known as Lewis-Sumner syndrome is clinically distinguished from typical CIDP due to its asymmetric multifocal sensory and motor symptoms [23]. Sensory CIDP includes chronic immune sensory polyradiculoneuropathy (CISP). This is differentiated from DADS as it is non-length dependent with normal sensory electro-diagnostic testing due to pathology of the dorsal root ganglion but abnormal somatosensory evoked potential. Cerebrospinal fluid (CSF) protein is elevated and MRI shows enlargement of nerve roots. CISP responds to typical CIDP treatment as noted below [24].
In addition, there is a subset of CIDP patients who present at a younger age, have a disabling tremor and are poorly responsive to the typical first line treatment of IVIg. These patients have been shown to have IgG4 antibodies against nodal and paranodal myelin-associated proteins—Contactin 1 and Neurofascin 155 [23].
Laboratory findings in CIDP patients include albumino-cytological dissociation in the CSF and abnormal electro-diagnostic study such as prolonged distal latency, slowed motor conduction velocity, prolonged F-waves, temporal dispersion and conduction block.
Involvement of T cells and B cells has been implicated in the pathogenesis of CIDP [7••]. T cell-mediated demyelination is thought to cause delamination of myelin lamellae. Induction of demyelination in animals by sera from patients of CIDP and immunoglobulin and complement deposits seen on nerve biopsies supports involvement of humoral immunity. Recently, IgG4 antibodies found against nodal and paranodal proteins Contactin 1 and Neurofascin 155 have an impact on diagnosis, prognosis and treatment [25]. The other proteins that may be implicated in the pathogenesis are gliomedin (it has a role in sodium channel clustering) or contactin-associated protein (CASPR1) [8••]. Some patients treated with PLEX or IVIg show improvement within days. This is not consistent with a process involving axon or myelin repair; thus, the reversal of antibody-mediated sodium channel alterations at the nodes of Ranvier induced by treatment is a possible explanation for this phenomenon [26]. See Figs. 1 and 2.
Treatment
Immunotherapy using IVIg, plasma exchange and/or corticosteroids is the first line of treatment for CIDP. The dose, duration and frequency of treatment with any of the above treatments are tailored to the patients’ response. There is no consensus regarding which first line treatment is the best. About 80–90% of patients improve with these treatments [27]. Patients that do not respond to the initial therapy should be switched to alternative therapy unless contraindicated.
IVIg was the first drug FDA approved in September 2008 for CIDP. The Immune Globulin Intravenous for Chronic Inflammatory Demyelinating Polyneuropathy (ICE) study showed that its benefit lasts for up to 48 weeks when given at maintenance dose of 1 g/kg every 3 weeks. It also showed that 8–12 weeks of treatment is required for maximal benefit [28]. Typically, IVIg is administered as an induction dose of 2 g/kg over 2–5 days followed by 0.5–2 g/kg every 2–6 weeks based on patients’ response. Eventually, the treatment goal is to reduce the frequency and taper IVIg completely after a few months of desired response. Though, the patients who can attain this remission are likely a small percentage.
Subcutaneous immunoglobulin (SCIg) was approved by the FDA in March 2018. It is convenient for the patient as it can be self-administered after appropriate training and is associated with lower healthcare costs compared with IVIg. The efficacy and tolerability of SCIg are similar to IVIg as found in the PATH study [29•]. SCIg, however takes longer to act compared with IVIg. Improvement of muscle strength peaked 2 weeks after IVIg, whereas it took 5 weeks after SCIg [30].
One unblinded randomised trial of 28 patients compared prednisone tapering from 120 mg every other day with no treatment which found that the patients in treatment arm improved, while those in untreated arm worsened over 3 months [31]. Other randomised control trials have compared the use of intravenous methylprednisolone with IVIg and oral daily prednisone with pulsed monthly dexamethasone [32, 33]. These studies showed no difference in efficacy; IVIg was better tolerated than corticosteroids. Typically, the steroid regimens used for CIDP are daily or alternate day oral prednisone at 1–1.5 g/kg followed by tapering over weeks to minimal required dose for response, or pulse weekly oral/intravenous methylprednisolone.
Two small randomised control trials have shown improved function in two-thirds of patients with CIDP treated with short-term PLEX compared with sham exchange [34]. Typically, PLEX is done in five sessions of 50 ml/kg plasma volume every other day followed by one to two sessions every 3–4 weeks depending upon response but is largely considered a second- or third-line therapy.
The second line of treatment for CIDP includes steroid sparing immunomodulatory therapy such as azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine and methotrexate.
Azathioprine, mycophenolate, cyclophosphamide and methotrexate are B and T lymphocyte inhibitors while cyclosporine is T cell inhibitor. Cyclophosphamide in pulsed moderate dosing as well as in high dose has the strongest evidence for use in refractory CIDP [35, 36]. Toxicity of this drug remains a limitation for its use. The limited data available for the use of these drugs does not show any significant benefit for the treatment of CIDP; however, none of these trials were large enough to rule out small or moderate benefit [37].
Rituximab which is a monoclonal antibody against CD20 protein of the B Lymphocytes has been used in patients with IgG4 anti-Contactin 1 and anti-Neurofascin 155 antibodies with benefit [38]. See Table 1.
Optimization of therapy
The response to treatment is gradual. Most patients who respond to IVIg need at least 3 maintenance infusions after 8–12 weeks of initiation of treatment while corticosteroids may need up to 12 weeks [28, 39]. Once maximum benefit has been achieved and sustained, reduction in frequency and dose should be attempted, while keeping in mind that this carries a risk of irreversible disability if relapse occurs. There is no standard approach to taper treatment and needs tailoring for individual patient. Objective assessment of patient’s response using standardised scales like Rasch-built overall disability scale (RODS), modified INCAT-sensory sum score (INCAT-SSS) and strength measurement using a grip strength dynamometer may provide a way to reduce the risk of early relapse and prevent over treatment [40, 41].
Rehabilitation
As mentioned above in GBS, physical and occupational rehabilitation is an important aspect of management of patients with CIDP.
Demyelinating neuropathies associated with monoclonal gammopathy
Neuropathies associated with paraproteinemias are heterogeneous clinically and electrophysiologically. In most cases, the pathologic significance of the paraprotein is unknown (MGUS). The monoclonal gammopathy can be of the IgG, IgA or IgM subtype. The non-IgM MGUS is the most common type, while IgM MGUS accounts for about 15% of MGUS cases.
Neuropathy associated with non-IgM MGUS
IgG and IgA paraproteins may be associated with neuropathies with clinical and electrophysiological features similar to CIDP as well as with distal axonal sensory or sensorimotor polyneuropathy [42].
Treatment
Treatment for patients with CIDP and non–IgM MGUS is the same as that for patients with CIDP without paraproteinemia [39]. For the distal axonal IgG/IgA MGUS, there is no evidence to support immunomodulatory treatment at this time. However, non IgM MGUS is considered to be pre-malignant, with 1% chance of malignant transformation, with a cumulative risk of 14% at 25 years. Hence, annual surveillance is recommended [43].
Neuropathy associated with IgM MGUS
Even though IgM paraprotein only forms about 20% of the gammopathies, 50% of patients with paraprotein and neuropathy have the IgM subtype [44]. Most patients with neuropathy associated with monoclonal gammopathy have MGUS; although, in some patients it may be associated with lymphoproliferative IgM secreting disorders such as Waldenstrom’s macroglobulinemia or lymphoma. Hence, it is essential to evaluate for these diseases.
Clinically, patients present with a distal acquired demyelinating symmetric neuropathy (DADS) phenotype with sensory ataxia due to involvement of large fibre sensory nerves and foot drop. They have significant large fibre dysfunction and distal weakness on exam. Many patients exhibit a kinetic tremor. Electro-diagnostically, they show distal demyelination [22].
Two-thirds of patients with IgM antibody have reactivity to neural antigens, most commonly to myelin-associated glycoprotein (MAG). MAG is a transmembrane glycoprotein concentrated in non-compacted periaxonal myelin regions. Anti-MAG neuropathy is usually seen in the sixth or seventh decades of life and affects men more than women. IgM anti-MAG antibodies are thought to be pathogenic as passive transfer of these human antibodies to animals results in neuropathy similar to that in humans. Also, IgM and complement deposits on the myelinated nerve fibres are seen on sural nerve biopsies [45].
Treatment
Patients with anti-MAG antibodies rarely respond to IVIg or steroids [46]. Rituximab has been shown to be helpful in 30–50% of patients with anti-MAG antibodies [47]. Multiple clinical and immunological biomarkers have been used to describe the characteristics of rituximab responders but it is still unclear [48••]. Demyelinating pattern, older age and absence of prior treatment seem significant risk factors for worsening disability. Gender, ataxia, tremor and IgM anti-MAG antibody titres are not helpful in predicting response [49], whereas motor deficits and subacute progression are factors predicting positive response [50].
The dose of rituximab used in studies is 375 mg/m2 every week for 4 weeks and may need maintenance doses every 12 weeks up to 2 years. Response to rituximab has led to interest in trying other novel and more potent anti-B cell agents. Ocrelizumab which is the humanised version of rituximab and induces more complement activation can be considered [48••]. Obinituzumab which is a humanised monoclonal antibody against CD 20 was used in two patients with anti-MAG neuropathy that did not respond to rituximab. The drug was found to be safe, but did not improve symptoms at 1 year of follow-up. It, however, normalised the IgM level and anti-MAG titres. This suggests that obinituzumab might be considered in refractory patients [51]. See Table 1.
POEMS syndrome
This syndrome consists of polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy (usually lambda) and skin abnormalities—hyperpigmentation, hypertrichosis, acrocyanosis, plethora and hemangioma/telangiectasia. At least three of these five features should be present to meet criteria for diagnosis. Polyneuropathy is a presenting feature of this syndrome in about 50% of patients. Clinically, they present similar to CIDP with distal neuropathy and severe pain. Osteoclastic plasmacytoma with sclerotic bone lesions (see Fig. 2 showing lytic bone lesion in a patient with POEMS syndrome) is present in most patients while some have monoclonal gammopathy of unknown significance (MGUS). Laboratory findings include markedly elevated CSF protein and serum vascular endothelial growth factor (VEGF). Electro-diagnostic testing shows a combination of severe axonal loss with diffusely slow velocities without any conduction block [52].
The pathogenesis of neuropathy in POEMS syndrome is related to overproduction of VEGF by monoclonal plasma cells which causes increased vascular permeability and neovascularization.
Treatment
This involves targeting the underlying plasma cell disorder. Focal osteoclastic lesions can be surgically excised or irradiated. Diffuse lesions require systemic therapy with a combination of corticosteroids and melphalan or high-dose chemotherapies and peripheral blood stem cell transplantation [53]. Other treatment options include bevacizumab, a monoclonal antibody against VEGF, and lenalidomide/thalidomide [54, 55].
Multifocal motor neuropathy with conduction block
Multifocal motor neuropathy with conduction block (MMN) is a rare condition affecting 1 in 100,000 individuals, with mean age of onset around 40 years with a male preponderance in the ratio of 3:1. It is a chronic, asymmetric and slowly progressive immune-mediated neuropathy. It typically first affects the forearm and intrinsic hand muscles and usually spares the sensory nerves [56]. Fasciculation and cramping can be seen in up to two-thirds of patients leading to a misdiagnosis of amyotrophic lateral sclerosis. Reflexes are usually absent or reduced in the affected regions [57]. Laboratory findings include presence of IgM antibodies to GM1 in about half the patients [58]. CSF protein is often normal or only mildly elevated. Electro-diagnostically, conduction block outside of typical compression sites seen in two or more nerves confirms the diagnosis but it is not essential as long as other typical features are present.
The immunopathology of MMN is uncertain. Traditionally, it has been thought to be a demyelinating disorder with secondary axonal involvement; however, early pathology at the nodes of Ranvier has now been implicated. IgM antibodies against GM1 are commonly found in patients with MMN. GM1 is found ubiquitously in sensory and motor nerves, but it is highly concentrated in the nodal and paranodal regions of the motor nerves [59]. Exposure to human sera containing anti-GM1 antibodies induces conduction block, immunoglobulin deposits at the nodes of Ranvier and nodal widening in animal nerve [60]. Reduction in sodium currents to eventually block action potential has been seen in the presence of GM1 antibodies and complement [61]. In humans with MMN, axonal hyperpolarisation has been seen at sites distal to conduction block [62]. The above observations imply that antibody-mediated complement injury at the nodes of Ranvier and adjacent axolemmal hyperpolarisation caused by sodium/potassium pump dysfunction mediate conduction block and resultant weakness [63]. This might also explain the clinical improvement within days seen in patients treated with IVIg due to reversal of antibody-mediated nodal dysfunction. See Fig. 1.
Treatment
IVIg is the first-line therapy for MMN. Most patients respond after the first dose of IVIg (2 g/kg over 2–5 days). Maintenance therapy is required in most patients, the dose and frequency of which are based on response. The typical dose being 1 g/kg every 2–4 weeks or 2 g/kg every 4–8 weeks. If treatment with IVIg is insufficient, other immune suppressive treatment may be considered; however, no agent has been shown to be effective in clinical trials and data from case series are conflicting. Use of cyclophosphamide is limited by its adverse effects [64]. Corticosteroids and plasma exchange have not been found effective and might worsen the disease [65]. See Table 1.
Two recent studies have looked at the use of SCIg in MMN. These have found different results, with one meta-analysis showing no difference in muscle strength in patients treated with IVIg and SCIg over 1–2 years while in the other study with CIDP and MMN patients treated with IVIg, it was seen that 40% of MMN patients required alterations in treatment regimen versus only 13% of CIDP patients requiring it to stabilise clinical condition [29•, 66]. Further studies are needed to assess long-term efficacy of SCIg in MMN. Complement activation has been found to be one of the pathogenic mechanisms of MMN. Hence, eculizumab could be a novel treatment option for MMN in the future.
Cancer-related neuropathies
This group of neuropathies can be classified as those related directly to the cancer or paraneoplastic neuropathies. The former can be from direct infiltration, compression and/or mass effect on the plexus or peripheral nerves. Another rare entity is neurolymphomatosis (NL), which involves infiltration of the cranial nerves, plexus, spinal nerve roots and/or peripheral nerves by the malignant lymphocytes. This condition is seen in patients with underlying leukaemia or non-Hodgkin’s lymphoma (NHL). Symptoms of NL include sensorimotor deficits, muscular atrophy, hypotonia, hyporeflexia and spontaneous pain. It appears that electro-diagnostic studies and positron emission tomography seem to be better diagnostic tools compared with spinal fluid analysis and MRI [67].
Treatment
Management primarily involves treating the underlying cancer. There is no standard treatment for NL but IVIg and/or PLEX have been tried in the past. More studies are necessary to evaluate the effectiveness of these therapies.
Paraneoplastic neuropathies
Paraneoplastic neuropathies can involve sensory or motor ganglia, peripheral sensory/motor nerves and autonomic nerves. Paraneoplastic sensory neuronopathy is one of the most common paraneoplastic syndromes [68]. This is associated with antibodies against type 1 antineuronal nuclear antigens (ANNA-1) previously called anti-Hu antibodies. Clinically, it is characterised by a subacute onset of sensory ataxia and neuropathic pain; concomitant motor neuropathy, cerebellar degeneration, limbic encephalitis and brainstem involvement [69]. Autonomic involvement causing orthostatic hypotension, cardiac arrhythmias and gastroparesis may also be seen. These antibodies react against neural intracellular antigens through cell-mediated immune mechanisms [70]. Small cell lung cancer is the most common underlying malignancy; others include carcinomas of the breast, ovaries, pancreas and lymphoma. Sensory neuronopathy may precede the diagnosis of cancer by up to 8 months or longer [71].
Less commonly, anti-CV2/collapsin response mediator protein-5 (CRMP 5) antibodies are associated with a mixed axonal and demyelinating motor polyneuropathy [72]. Isolated paraneoplastic autonomic neuropathy can be seen in association with antibodies to ganglionic nicotinic acetylcholine receptor [73]. Symptomatic chronic sensorimotor polyneuropathy is seen in 10–15% of patients with solid tumours, especially lung cancers and some have subclinical evidence of the same. Acute sensorimotor and demyelinating neuropathies which resemble GBS or CIDP have been seen in hematologic malignancies, most commonly Hodgkin’s lymphoma. Isolated paraneoplastic motor neuropathy or neuronopathy which mimic amyotrophic lateral sclerosis has rarely been reported [74].
Treatment
These are rare disorders; hence, recommendations are mostly based on expert opinion. Management is broadly divided into identification and treatment of underlying malignancy, immunosuppression/immunomodulation and symptomatic treatment. Treatment of the cancer may stabilise or perhaps improve the paraneoplastic syndromes associated with intracellular antigens [75]. Corticosteroids, cyclophosphamide, sirolimus and rituximab are the immunosuppressants used to treat paraneoplastic sensory neuronopathy. Immunomodulation using IVIg and plasma exchange can be used [71, 74]. Symptomatic treatment involves use of neuropathic medications for pain. See Table 1.
Patients presenting with an AIDP- or CIDP-like symptoms are treated similar to a patient without cancer—IVIg or plasma exchange for the former and corticosteroids or IVIg for the latter.
Vasculitic neuropathy
This group of neuropathy which results from inflammation of vasa nervosum, is asymmetric, painful, abrupt and multifocal in nature involving the sensory and motor nerves [76]. It often progresses quickly over hours to days, in a stepwise manner in the distribution of individual nerves causing pain, weakness and numbness. Systemic symptoms like weight loss, myalgia, fatigue and fever are also present. Vasculitis also affects other organ systems like the skin, kidneys, liver, heart, brain and lungs causing ischemic injury to them. This usually occurs in the setting of a systemic disease that predisposes to vasculitis. These include infections like hepatitis C-associated cryoglobulinemia, connective tissue disorders like rheumatoid arthritis or systemic lupus erythematosus, inflammatory conditions like Wegener’s granulomatosis, Churg-Strauss angiitis, polyarteritis nodosa or microscopic polyangiitis and paraneoplastic syndromes [76]. About 10–15% patients have non-systemic vasculitic neuropathy. Laboratory studies show markers for the underlying disease and electro-diagnostic testing shows axonal sensory and motor neuropathy in multiple affected nerves. T cells play a role in the pathogenesis.
Treatment
There are no randomised clinical trials so far. Emergent immunosuppressive treatment is needed to avoid the various systemic complications related to ischemia. Steroids in combination with steroid sparing agents are traditionally used [77]. Initially, prednisone 40–100 mg daily is used for 6–8 weeks or until treatment response and is followed by a gradual taper to alternate days. Prednisone is continued for 6 months. In severe cases, cyclophosphamide is given in addition to steroids. It is administered either orally (1 mg/kg daily) or intravenously (1000 mg/m2 monthly) for 6 months. Therapy may need to be continued for a year or longer [69]. IVIg and plasma exchange may also be used [78]. Rituximab (375 mg/m2 every week for 4 weeks) may also be an alternative in patients with intolerable side effects from cyclophosphamide [79]. In patients with hepatitis C, antiviral therapy using nucleotide analog inhibitors is used. See Table 1.
Drug-induced immune-mediated neuropathies
This is a small and rare group of neuropathies and includes immune-mediated neuropathies caused by TNF alpha inhibitors and newer anti-cancer drugs.
There are several aetiologies of neuropathy in cancer patients; often, it is due to toxicity of chemotherapeutic agents. Less commonly it can be paraneoplastic, immune mediated or neoplastic neuropathy. Immune checkpoint inhibitors are a relatively newer group of anti-cancer drugs used in the treatment of melanoma and other tumours. These are monoclonal antibodies that attack the human cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and include the drugs ipilimumab and tremelimumab. There have been reports about ipilimumab causing a CIDP-like illness [80]. The other group of immune checkpoint inhibitors target the programmed cell death 1 (PD-1) receptor, which regulates cell death in immune cells. This includes pembrolizumab and nivolumab. AIDP and CIDP have been reported with these drugs [81]. Treatment of these side effects includes discontinuing the offending drug and administration of immunotherapy (steroids and/ or IVIg), which has been reported to be beneficial.
The other group of drugs that have been associated with immune neuropathies are tumour necrosis factor alpha antagonists used in the treatment of inflammatory conditions including rheumatoid arthritis, spondyloarthritis, psoriasis and inflammatory bowel disease. There have been case reports about infliximab being associated with rare neurological complications including GBS, CIDP and MMN [82, 83]. The pathogenesis is thought to be from T cell and humoral attack against peripheral nerve myelin, vasculitis-induced nerve ischemia and inhibition of signalling support for axons [84]. The management involves stopping the offending drug as the first step which leads to resolution of symptoms [85]. Corticosteroids, IVIg and plasma exchange can also be used [84].
Future direction
In spite of important advances in the discovery of immunopathogenesis, diagnosis and treatment of immune-mediated neuropathies, there are several questions that need to be answered for better management of patients, especially those with refractory disease that do not respond to the standard treatment. The discovery of pathogenic antibodies to nodal and paranodal proteins in a subset of patients with CIDP has helped in providing a tailored treatment to them with better outcomes. Similarly, discovery of more specific biomarkers and better clinical criteria would help towards accurate diagnosis. This would further pave the way for personalised medicine in immune-mediated neuropathies, wherein treatment can be individualised to each patient.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Barohn RJ. Approach to peripheral neuropathy and neuronopathy. Semin Neurol. 1998;18:7–18.
Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet. 2016;388:717–27.
•• Esposito S, Longo MR. Guillain-Barre syndrome. Autoimmun Rev. 2017;16:96–101. This review summarises the clinical features and diagnostic criteria of GBS and proposes the algorithm for management.
Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27 Suppl:S21–4.
van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–82.
Ropper AH. The Guillain-Barré syndrome. N Engl J Med. 1992;326:1130–6.
•• Dalakas MC. Pathogenesis of immune-mediated neuropathies. Biochim Biophys Acta. 2015;1852:658–66. This article reviews the pathogenesis of GBS, CIDP, MMN and anti-MAG neuropathy.
•• Kieseier BC, Mathey EK, Sommer C, Hartung H-P. Immune-mediated neuropathies. Nat Rev Dis Prim. 2018;4:31. This is a recent comprehensive review paper on pathogenesis, diagnosis and management of AIDP, CIDP and MMN.
Lawn ND, Fletcher DD, Henderson RD, Wolter TD, Wijdicks EF. Anticipating mechanical ventilation in Guillain-Barre syndrome. Arch Neurol. 2001;58:893–8.
Ruts L, Drenthen J, Jongen JLM, Hop WCJ, Visser GH, Jacobs BC, et al. Pain in Guillain-Barre syndrome: a long-term follow-up study. Neurology. 2010;75:1439–47.
The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barre syndrome. Neurology. 1985;35:1096 LP–1096.
van der Meché FGA, Schmitz PIM. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;326:1123–9.
Hughes RAC, Swan A V, van Doorn PA. Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2014;CD002063.
Hughes RAC, Swan AV, Raphaël J-C, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130:2245–57.
Hughes RA. Plasma exchange versus intravenous immunoglobulin for Guillain-Barré syndrome. Ther Apher. 1997;1:129–30.
Hughes RAC, Brassington R, Gunn AA, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2016.
Davidson AI, Halstead SK, Goodfellow JA, Chavada G, Mallik A, Overell J, et al. Inhibition of complement in Guillain-Barre syndrome: the ICA-GBS study. J Peripher Nerv Syst. 2017;22:4–12.
Misawa S, Kuwabara S, Sato Y, Yamaguchi N, Nagashima K, Katayama K, et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: a multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018;17:519–29.
Herbison GJ, Jaweed MM, Ditunno JFJ. Exercise therapies in peripheral neuropathies. Arch Phys Med Rehabil. 1983;64:201–5.
Lunn MPT, Manji H, Choudhary PP, Hughes RAC, Thomas PK. Chronic inflammatory demyelinating polyradiculoneuropathy: a prevalence study in south east England. J Neurol Neurosurg Psychiatry. 1999;66:677–80.
Mathey EK, Park SB, Hughes RAC, Pollard JD, Armati PJ, Barnett MH, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86:973–85.
Katz JS, Saperstein DS, Gronseth G, Amato AA, Barohn RJ. Distal acquired demyelinating symmetric neuropathy. Neurology. 2000;54:615–20.
Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958–64.
Sinnreich M, Klein CJ, Daube JR, Engelstad J, Spinner RJ, Dyck PJB. Chronic immune sensory polyradiculopathy. Neurology. 2004;63:1662–9.
Querol L, Illa I. Paranodal and other autoantibodies in chronic inflammatory neuropathies. Curr Opin Neurol. 2015;28:474–9.
Berger M, Allen JA. Optimizing IgG therapy in chronic autoimmune neuropathies: a hypothesis driven approach. Muscle Nerve. 2015;51:315–26.
Cocito D, Paolasso I, Antonini G, Benedetti L, Briani C, Comi C, et al. A nationwide retrospective analysis on the effect of immune therapies in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2010;17:289–94.
Hughes RAC, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–44.
• van Schaik IN, Bril V, van Geloven N, Hartung H-P, Lewis RA, Sobue G, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2018;17:35–46 Study on SCIg in CIDP.
Markvardsen LH, Sindrup SH, Christiansen I, Olsen NK, Jakobsen J, Andersen H. Subcutaneous immunoglobulin as first-line therapy in treatment-naive patients with chronic inflammatory demyelinating polyneuropathy: randomized controlled trial study. Eur J Neurol. 2017;24:412–8.
Dyck PJ, O’Brien PC, Oviatt KF, Dinapoli RP, Daube JR, Bartleson JD, et al. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann Neurol. 1982;11:136–41.
Nobile-Orazio E, Cocito D, Jann S, Uncini A, Beghi E, Messina P, et al. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: a randomised controlled trial. Lancet Neurol. 2012;11:493–502.
van Schaik IN, Eftimov F, van Doorn PA, Brusse E, van den Berg LH, van der Pol WL, et al. Pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy (PREDICT study): a double-blind, randomised, controlled trial. Lancet Neurol. 2010;9:245–53.
Mehndiratta MM, Hughes RAC, Pritchard J. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2015;CD003906.
Good JL, Chehrenama M, Mayer RF, Koski CL. Pulse cyclophosphamide therapy in chronic inflammatory demyelinating polyneuropathy. Neurology. 1998;51:1735 LP–1738.
Brannagan TH 3rd, Pradhan A, Heiman-Patterson T, Winkelman AC, Styler MJ, Topolsky DL, et al. High-dose cyclophosphamide without stem-cell rescue for refractory CIDP. Neurology. 2002;58:1856–8.
Mahdi-Rogers M, Brassington R, Gunn AA, van Doorn PA, Hughes RA. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2017;5:CD003280.
Querol L, Rojas-Garcia R, Diaz-Manera J, Barcena J, Pardo J, Ortega-Moreno A, et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflammation. 2015;2:e149.
Van den Bergh PYK, Hadden RDM, Bouche P, Cornblath DR, Hahn A, Illa I, et al. European Federation of Neurological Societies/peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripher. Eur J Neurol. 2010;17:356–63.
van Nes SI, Vanhoutte EK, van Doorn PA, Hermans M, Bakkers M, Kuitwaard K, et al. Rasch-built overall disability scale (R-ODS) for immune-mediated peripheral neuropathies. Neurology. 2011;76:337–45.
Draak THP, Pruppers MHJ, van Nes SI, Vanhoutte EK, Bakkers M, Gorson KC, et al. Grip strength comparison in immune-mediated neuropathies: vigorimeter vs. Jamar. J Peripher Nerv Syst. 2015;20:269–76.
Magy L, Chassande B, Maisonobe T, Bouche P, Vallat J-M, Leger J-M. Polyneuropathy associated with IgG/IgA monoclonal gammopathy: a clinical and electrophysiological study of 15 cases. Eur J Neurol. 2003;10:677–85.
Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346:564–9.
Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362–9.
Tatum AH. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann Neurol. 1993;33:502–6.
Dalakas MC. Pathogenesis and treatment of anti-MAG neuropathy. Curr Treat Options Neurol. 2010;12:71–83.
Dalakas MC. Rituximab in anti-MAG neuropathy: more evidence for efficacy and more predictive factors. J Neurol Sci. 2017;377:224–6.
•• Dalakas MC. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther Adv Neurol Disord. 2018;11:1756285617746640. Detailed review on anti-MAG neuropathy.
Galassi G, Tondelli M, Ariatti A, Benuzzi F, Nichelli P, Valzania F. Long-term disability and prognostic factors in polyneuropathy associated with anti-myelin-associated glycoprotein (MAG) antibodies. Int J Neurosci. 2017;127:439–47.
Gazzola S, Delmont E, Franques J, Boucraut J, Salort-Campana E, Verschueren A, et al. Predictive factors of efficacy of rituximab in patients with anti-MAG neuropathy. J Neurol Sci. 2017;377:144–8.
Rakocevic G, Outschoorn UM, Dalakas M. Obinutuzumab (GAZYVA), a potent anti-B cell agent, in the treatment of rituximab-unresponsive IgM anti-myelin-associated-glycoprotein (MAG)-mediated-neuropathy (P1.450). Neurology. 2018;90:P1.450.
Mauermann ML, Sorenson EJ, Dispenzieri A, Mandrekar J, Suarez GA, Dyck PJ, et al. Uniform demyelination and more severe axonal loss distinguish POEMS syndrome from CIDP. J Neurol Neurosurg Psychiatry. 2012;83:480–6.
Li J, Zhang W, Jiao L, Duan M-H, Guan H-Z, Zhu W-G, et al. Combination of melphalan and dexamethasone for patients with newly diagnosed POEMS syndrome. Blood. 2011;117:6445–9.
Badros A, Porter N, Zimrin A. Bevacizumab therapy for POEMS syndrome. Blood. 2005;106:1135.
Chu BF, Shana’ah A, Hofmeister CC, Benson DM, Sell M, Tucker J, et al. Long-term therapy with lenalidomide in a patient with POEMS syndrome. Eur J Case Rep Intern Med. 2014;1:2014_000093.
Guimaraes-Costa R, Bombelli F, Leger J-M. Multifocal motor neuropathy. Curr Opin Neurol. 2013;26:503–9.
Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol. 2000;57:109–13.
Vlam L, van den Berg LH, Cats EA, Piepers S, van der Pol W-L. Immune pathogenesis and treatment of multifocal motor neuropathy. J Clin Immunol. 2013;33(Suppl 1):S38–42.
Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–625.
Uncini A, Santoro M, Corbo M, Lugaresi A, Latov N. Conduction abnormalities induced by sera of patients with multifocal motor neuropathy and anti-GM1 antibodies. Muscle Nerve. 1993;16:610–5.
Takigawa T, Yasuda H, Kikkawa R, Shigeta Y, Saida T, Kitasato H. Antibodies against GM1 ganglioside affect K+ and Na+ currents in isolated rat myelinated nerve fibers. Ann Neurol. 1995;37:436–42.
Kiernan MC, Guglielmi J, Kaji R, Murray NMF, Bostock H. Evidence for axonal membrane hyperpolarization in multifocal motor neuropathy with conduction block. Brain. 2002;125:664–75.
Uncini A, Kuwabara S. Nodopathies of the peripheral nerve: an emerging concept. J Neurol Neurosurg Psychiatry. 2015;86:1186–95.
van Schaik IN, Léger JM, Nobile-Orazio E, Cornblath DR, Hadden RD, Koski CL, et al. European Federation of Neurological Societies/peripheral nerve society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society--first revisi. J Peripher Nerv Syst. 2010;15:295–301.
Carpo M, Cappellari A, Mora G, Pedotti R, Barbieri S, Scarlato G, et al. Deterioration of multifocal motor neuropathy after plasma exchange. Neurology. 1998;50:1480–2.
Cocito D, Merola A, Romagnolo A, Peci E, Toscano A, Mazzeo A, et al. Subcutaneous immunoglobulin in CIDP and MMN: a different long-term clinical response? J Neurol Neurosurg Psychiatry. 2016;87:791–3.
Kamiya-Matsuoka C, Shroff S, Gildersleeve K, Hormozdi B, Manning JT, Woodman KH. Neurolymphomatosis: a case series of clinical manifestations, treatments, and outcomes. J Neurol Sci. 2014;343:144–8.
Giometto B, Grisold W, Vitaliani R, Graus F, Honnorat J, Bertolini G. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol. 2010;67:330–5.
Gwathmey KG. Sensory neuronopathies. Muscle Nerve. 2016;53:8–19.
Darnell RB. Paraneoplastic neurologic disorders: windows into neuronal function and tumor immunity. Arch Neurol. 2004;61:30–2.
Rudnicki SA, Dalmau J. Paraneoplastic syndromes of the peripheral nerves. Curr Opin Neurol. 2005;18:598–603.
Antoine JC, Honnorat J, Camdessanche JP, Magistris M, Absi L, Mosnier JF, et al. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 2001;49:214–21.
Muppidi S, Vernino S. Paraneoplastic neuropathies. Continuum (Minneap Minn). 2014;20:1359–72.
Koike H, Sobue G. Paraneoplastic neuropathy. Handb Clin Neurol. 2013;115:713–26.
Graus F, Keime-Guibert F, Reñe R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–48.
Gwathmey KG, Burns TM, Collins MP, Dyck PJB. Vasculitic neuropathies. Lancet Neurol. 2014;13:67–82.
Gorson KC. Therapy for vasculitic neuropathies. Curr Treat Options Neurol. 2006;8:105–17.
Levy Y, Uziel Y, Zandman GG, Amital H, Sherer Y, Langevitz P, et al. Intravenous immunoglobulins in peripheral neuropathy associated with vasculitis. Ann Rheum Dis. 2003;62:1221–3.
De Vita S, Quartuccio L, Isola M, Mazzaro C, Scaini P, Lenzi M, et al. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheum. 2012;64:843–53.
Thaipisuttikul I, Chapman P, Avila EK. Peripheral neuropathy associated with ipilimumab: a report of 2 cases. J Immunother. 2015;38:77–9.
de Maleissye M-F, Nicolas G, Saiag P. Pembrolizumab-induced demyelinating polyradiculoneuropathy. N Engl J Med. 2016;375:296–7.
Uyanikoglu A, Ermis F, Akyuz F, Pinarbasi B, Baran B, Aydogan T, et al. Infliximab in inflammatory bowel disease: attention to adverse events. Eur Rev Med Pharmacol Sci. 2014;18:2337–42.
Seo B, Jeong YJ, Hong S, Kim Y-G, Lee C-K, Yoo B. A case of infliximab-induced multifocal motor neuropathy in a patient with rheumatoid arthritis and literature review. J Rheum Dis. 2016;23:250–5.
Stubgen J-P. Tumor necrosis factor-alpha antagonists and neuropathy. Muscle Nerve. 2008;37:281–92.
Kaltsonoudis E, Zikou AK, Voulgari PV, Konitsiotis S, Argyropoulou MI, Drosos AA. Neurological adverse events in patients receiving anti-TNF therapy: a prospective imaging and electrophysiological study. Arthritis Res Ther. 2014;16:R125.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Mithila Fadia and Sheetal Shroff each declare no potential conflicts of interest. Ericka Simpson has served on the speaker bureau for indications for subcutaneous IgG in CIDP patients sponsored by CSL Behring, Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuromuscular Disorders
Rights and permissions
About this article

Cite this article
Fadia, M., Shroff, S. & Simpson, E. Immune-Mediated Neuropathies. Curr Treat Options Neurol 21, 28 (2019). https://doi.org/10.1007/s11940-019-0569-y
Published:
DOI: https://doi.org/10.1007/s11940-019-0569-y