
Opinion statement
The management of patients with large territory ischemic strokes and the subsequent development of malignant brain edema and increased intracranial pressure is a significant challenge in modern neurology and neurocritical care. These patients are at high risk of subsequent neurologic decline and are best cared for in an intensive care unit or a comprehensive stroke center with access to neurosurgical support. Risks include hemorrhagic conversion, herniation, poor functional outcome, and death. This review discusses recent advances in understanding the pathophysiology of edema formation, identifying patients at risk, current management strategies, and emerging therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
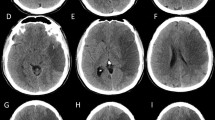
Stroke is a major cause of mortality, and it is estimated that large hemispheric strokes account for up to 10 % of all stroke patients [1, 2]. The term “malignant” was first used by Hacke et al. [3] to describe a complete infarction of the middle cerebral artery (MCA) accompanied by space-occupying mass effect within the first 5 days after presentation. Malignant cerebral infarction (MCI) typically refers to large MCA territory infarction (with or without involvement of the ipsilateral anterior or posterior cerebral artery territories) that presents with acute brain swelling in the first 48 h after ictus. The resultant tissue shifts secondary to regional changes in intracranial pressure (ICP) may lead to brain herniation [4]. The mortality rate in MCI can be as high as 80 %, usually following transtentorial herniation after development of brain edema [2, 3]. In contrast, the average mortality rate among all ischemic stroke subtypes is roughly 15 %, with MCA territory infarcts in general ranging from 5 %‒45 % [1, 5]. Concurrent anterior cerebral artery (ACA) involvement may be predictive of mortality [6]. Morbidity has not been as well documented but few patients with MCI are likely to ever return to independent functioning [7].
Subtotal or complete MCA territory infarctions account for approximately 10 % of anterior circulation stroke patients [8]. Etiology may be thrombotic, embolic, or secondary to an internal carotid artery (ICA) dissection with the occlusion typically involving the ICA or proximal MCA [9]. The size of the resultant infarct depends on the vessel that is occluded and the extent of collateral circulation available to the ischemic territory.
Patients with proximal MCA or ICA occlusion present awake with contralateral hemiparesis and sensory deficit affecting face and arm with variable leg involvement. Gaze deviation toward the side of the injury and contralateral hemineglect are common. Dominant hemisphere infarction presents with a global aphasia. National Institutes of Health Stroke Score is commonly 20 or higher for dominant hemisphere infarcts and up to 15 or more for nondominant hemisphere infarcts [10]. New onset anisocoria or hemiplegia ipsilateral to stoke should be taken as evidence of transtentorial herniation until proven otherwise and should be evaluated and treated emergently. Decreased level of consciousness may signal worsening midline shift (notably at the level of the pineal gland) [11] or thalamic or midbrain compression [12]. Patients with hemispheric strokes may also develop “eye lid apraxia” or “cerebral ptosis,” which is oftentimes mistaken for decreased consciousness [13].
Similar to patients with large hemispheric strokes, patients with cerebellar strokes are also at risk for neurologic deterioration because of development of edema. Cerebellar stroke may be more difficult to recognize because of nonspecific complaints (eg, nausea, dizziness, vertigo) and may be missed without careful evaluation of speech, eye movements, gait, and coordination. Swelling may result in pontine compression and/or acute hydrocephalus because of compression of the fourth ventricle. Unilateral hearing loss may be detected in anterior inferior cerebellar artery infarcts whereas intractable singultus may occur in posterior inferior cerebellar artery infarcts [14]. Subsequent deterioration depends on the size of the initial infarct volume [15]. The initial computed tomography scan, however, may be normal in up to 25 % of patients [15, 16]. In addition to decreased consciousness, patients with pontine compression may develop ophthalmoparesis, disordered breathing, or cardiac dysrhythmias.
Historically, treatment of large strokes has focused on symptom management, however, multicenter trials are now underway examining potential interventions to prevent the development of edema.
Mechanism of edema formation
Dysfunction of cerebral capillaries because of ischemia and postischemic reperfusion results in progressive alterations in blood-brain barrier (BBB) leading to formation of ionic edema, vasogenic edema, and hemorrhagic conversion [17]. Ischemic injury leads to altered ion gradients primarily through cessation of active transport Na-K-ATPase channels. Passive ion transporters then allow intracellular accumulation of sodium in neurons and glial cells resulting in cellular swelling. This is classically termed “cytotoxic edema” [18•]. Hyperintense signal on diffusion weighted magnetic resonance imaging (MRI) results from this process. Endothelial cell dysfunction follows with subsequent disruption of the BBB. Vasogenic edema then develops as fluid leaks from the intravascular space. Patients are at increased risk of hemorrhagic transformation after the development of vasogenic edema because of increased permeability of the BBB [17].
Several ion channels involved in edema formation are being studied as potential therapeutic targets. The SUR1/TRPM4 is activated and upregulated following ischemia. This results in an influx of cations and cellular swelling. SUR1/TRMP4 channel activation also leads to an upregulation of matrix-metalloproteinase-9 (MMP-9) [19]. MMP-9 is associated with endothelial damage and increased vascular permeability. NKCC1, an active transported involved with the Na-K-Cl channel mediating sodium entry into cells, is activated following ischemic injury [20]. Aquaporin-4, the most abundant water channel in the brain, is also known to be involved in edema formation after stroke [21].
Predictors of malignant cerebral edema
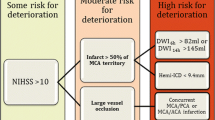
Numerous clinical and radiographic predictors of malignant cerebral infarction have been studied, though no single marker can be relied on exclusively. A 2008 systematic review of 23 published studies found that the most significant predictors were infarct size, involvement of vascular territories in addition to the MCA, low perfusion in other vascular territories, and hemorrhagic transformation [22]. Clinical characteristics that have been associated with a malignant course include younger age [9], female sex [9, 23], systolic blood pressure >180 mm Hg [24] or history of hypertension [25], congestive heart failure[25], coronary artery disease [26], leukocytosis [25], or National Institutes of Health Stroke Score severity [27, 28]. Serum protein S-100 [29], cellular fibronectin, and MMP-9 [30] are promising predictive biomarkers, though at present their clinical usefulness has not been established.
Computed tomography predictors identified include hypodensity >50 % of MCA territory on initial computed tomography [24, 25, 31], pineal displacement >4 mm within 48 h of onset [32], internal carotid artery T occlusion [28, 33], combined ICA and MCA occlusion [34], involvement of vascular territory other than MCA (eg, ACA, PCA, anterior choroidal) [25, 23], hyperdense MCA sign [31, 35], attenuated corticomedullary contrast within 18 h of onset [35], and perfusion deficit in >66 % of MCA territory at 6 h after onset [36]. MRI predictors include apparent diffusion coefficient (<80 %) [28] or diffusion weighted imaging [34] volume lesion volume >82 mL within 6 h of onset, and time-to-peak on MRI perfusion maps >162 mL [28].
Treatment
Basic support
-
Patients that have large territorial strokes should be admitted to a neuro-intensive care unit or stroke unit for close monitoring. Transfer to a higher level center is reasonable if comprehensive care and neurosurgical intervention are not available locally. Endotracheal intubation may be considered in patients with decreased level of consciousness leading to hypoxia or impaired ability to maintain a patent airway. Maintaining normocarbia is reasonable, but prophylactic hyperventilation is not recommended [37].
-
New or worsening cardiac dysrhythmias are not uncommon after large ischemic stroke, especially in patients with cerebellar strokes leading to brainstem compression or strokes involving the insular region [38]. Continuous cardiac monitoring and pharmaceutical management of arrhythmias is recommended. There is insufficient data to recommend specific systolic or mean arterial pressures, as blood pressure goals will be influenced by a number of factors including whether or not the patient received thrombolytics or endovascular treatment for recanalization. However, marked hypertension (SBP >220 mm Hg or DBP >105 mm Hg) is associated with increased risk of hemorrhagic conversion [39]. Blood pressure targets will likely be lower following thrombolytics or intervention. Labetalol, hydralazine, or enalapril can be considered as needed. If continuous intravenous drip is needed, nicardipine can be titrated to blood pressure goal typically with a range of 5‒20 mg/h. Nitroprusside is generally avoided in patients with brain injury as it may theoretically increase ICP because of cerebral vasodilation [7].
-
Hyperglycemia is associated with increased infarct size, worsening cerebral edema, and increased risk of hemorrhagic transformation [40]. Hypoglycemia should be avoided and a recent trial demonstrated increased infarct size with aggressive glucose control (<126 mg/dL) [41]. Current recommendations are for target blood glucose between 140 and 180 mg/dL.
-
Aggressive treatment of fever is reasonable as hyperthermia has been shown to increase infarct size in animal models [42] and to worsen outcomes in neurological critical care populations [43]. Fever should prompt evaluation for infectious or drug-induced cause. Aspiration pneumonia is common. Patients should have their swallow function evaluated and be placed in semirecumbant position to reduce aspiration risk [44]. Most will likely need nasogastric tube placement for initial nutrition.
ICP monitoring
-
The 2013 Guidelines for early management of patients with acute ischemic stroke recommend similar initial interventions as those used in traumatic brain injury for the management of elevated ICP: hyperventilation, osmotic therapy, intraventricular cerebrospinal fluid drainage, and surgical decompression [45]. There, however, is no evidence that hyperventilation, osmotic therapy, corticoseteroids, diuretics or glycerol, or other measures that lower ICP alone improve outcomes in patients ischemic brain swelling [45].
-
Over the last several decades, it has become increasingly apparent that regional ICP differentials leading to displacement of midline structures (eg, thalamus or brainstem) are more often the cause of clinical deterioration than global elevations in ICP expected to result in downward displacement. As hypoventilation and osmotic therapy work primarily on healthy brain, this raised concerns that these types of therapies could worsen midline shift by shrinking noninfarcted brain contralateral and leaking into and expanding the edematous lesion [46]. While mannitol boluses have been shown to preferentially shrink noninfarcted brain, this has not appeared to be sufficient to worsen midline shift [47, 48].
-
There does not appear to be value in early ICP monitoring or ventriculostomy in hemispheric MCI [49]. In patients with cerebellar infarct with early edema, acute hydrocephalus may develop. Ventriculostomy is usually accompanied by suboccipital decompression [50].
Osmotic therapy
-
Mannitol and hypertonic saline are the most commonly used osmotic agents to reduce ICP. The acute effects of osmotic agents probably work through rheological effects drawing water out of brain cells into the intravascular space, decreasing blood viscosity, and reducing cerebral blood volume [51]. However, a recent study failed to demonstrate that either mannitol or hypertonic saline reduced cerebral blood volume [52].
-
Mannitol for symptomatic brain edema should be dosed at 1 g/kg repeated every 4‒6 h [53]. Mannitol has the advantage of being able to be given through peripheral intravenous access. Potential complications include hypovolemia, hypotension, and nephrotoxicity. It is common to monitor serum osmolality and withhold doses if osmolality is greater than 320 mOsm, however, there is no data to support this limit. Monitoring of the osmolar gap may be a more appropriate target, with a closing of the gap indicating sufficient mannitol clearance for additional dosing [54]. A recent Cochrane review did not find sufficient support for its routine use in stroke patients, however, only three studies were eligible for analysis [55].
-
Hypertonic saline is available in varying concentrations from 1.5 % to 23.4 % with concentrations of 3 % or higher requiring central venous access to avoid sclerosis of peripheral veins [56]. Rapid elevation of sodium in this context does not appear to be associated with complications of rapid sodium correction in hyponatremic patients (eg, osmotic demyelination). Hypertonic saline works on both injured and uninjured brain leading some to speculate it may be superior to mannitol [57]. Serum sodium levels up to 160 mmol/L may be appropriate, though levels above that are likely associated with increased complications including delirium and renal toxicity. Hypertonic saline may worsen congestive heart failure.
-
There is limited data comparing mannitol and hypertonic saline. Similarly, it is common to use these medications in recurrent boluses or continuous infusion in the case of hypertonic saline without high quality evidence. As osmotic gradients will equilibrate over time the benefits of osmotic therapies would be expected to be transient [58]. Similarly, discontinuation of hyperosmotic therapy may result in transient swelling of cells [59]. Data for a “rebound” edema is currently lacking.
Surgical decompression
-
Decompressive surgery is the current definitive treatment for malignant cerebral edema. The benefit of suboccipital decompression for patients with neurologic deterioration after cerebellar stroke is well established. In a large series of massive cerebellar infarcts, 40 % required surgical decompression while 17 % were managed with ventriculostomy; 74 % of patients in this series survived with modified Rankin scores (mRS) of 0 or 1 (able to carry out all previous activities) [60].
-
Decompressive craniectomy for large hemispheric infarction has become more common following publication of a pooled analysis [61] of three multiple randomized controlled trials in 2007. The DECIMAL (decompressive craniectomy in malignant middle cerebral artery infarcts) trial [62], DESTINY (decompressive surgery for the treatment of maligant infarction of the middle cerebral artery) trial[63], and HAMLET (hemicraniectomy after middle cerebral infarction with life-threatening edema trial) [64] trials enrolled patients aged 18‒60. The individual studies showed reduced mortality but none of them showed improvement in functional outcomes (defined as a mRS ≤3) compared with medical therapy. In the pooled analysis, there was a benefit in mortality (78 % vs 29 %), mRS ≤4 (75 % vs 24 %), and mRS ≤3 (43 % vs 21 %). The NNT for a mRS ≤3 was 4. In the pooled analysis, 55 % of survivors had mRS 3 or less and 45 % had moderate or severe disability at 12 months.
-
Two randomized controlled trials have examined decompressive craniectomy in patients older than 60 years of age. Both the study by Zhao et al [65] and the Destiny-II [66] trials demonstrated a mortality benefit and mRS <5. A potential criticism has been that survivors may have similar functional outcomes (mortality benefit without disability benefit).
-
The optimum clinical, radiographic, and timing cut-offs for surgery remains controversial. Early decompression may lead to some unnecessary surgeries while waiting for clinical deterioration may increase the likelihood for a poor outcome. It may be possible to develop protocols that distinguish high risk from low risk patients as evidenced by the HeADDFIRST (Hemicraniectomy and Durotomy Upon Deterioration From Infarction-Related Swelling Trial) trial [67]. The side of infarct should probably not affect the decision to pursue hemicraniectomy [68].
-
The major hemicranectomy trials all utilized creation of a large frontotemporoparietal bone flap followed by durotomy and duroplasty. A bone flap of at least 14 cm is required [69]. Postoperative complications include wound dehiscence among others. The optimum timing of cranioplasty is decompressive hemicraniectomy is unknown. It is common for patients to also require tracheostomy and gastrostomy [37].
Neuroprotection
Corticosteroids
-
Corticosteroids are not currently recommended for treatment of malignant cerebral edema in stroke. A 2012 Cochrane review [70] did not find evidence for survival benefit or improvement in functional outcomes in survivors. The trials included were small, underpowered, and heterogeneous in their outcome measurements.
Barbiturates
-
Barbiturates are effective in lowering ICP by lowering metabolic rate and may have neuroprotective effects as free radical scavengers [71]. However, barbiturate coma to lower ICP in large hemispheric strokes has not been shown to improve outcome [72]. Barbiturates are not currently recommended for use in ischemic cerebral or cerebellar edema.
Therapeutic hypothermia
-
Body temperature has been shown to influence outcomes in focal ischemic stroke experimental models and acute stroke in humans. Hyperthermia is associated with worse outcomes [73, 74], whereas a body temperature below 36.5 °C has been associated with improved outcome and lower mortality [75]. At present, there is insufficient data to recommend therapeutic hypothermia. Prospective randomized trials are currently in process including combining endovascular cooling with acute thrombolysis (ICTuS 2/3, clinicaltrials.gov NCT01123161) [76].
Glyburide
-
Glyburide (a.k.a glibenclamide) is a selective inhibitor of the sulfonylurea receptor 1 (SUR1) on the SUR1/TRPM4 channel. Inhibition of this receptor may limit edema formation by reducing MMP-9 activation. Retrospective studies have shown that diabetic patients continued on their pre-stroke sulfonylurea after hospitalization for acute ischemic stroke had improved functional outcomes [77] and reduced risk of hemorrhagic conversion [78]. A recent pilot study demonstrated reduced MRI evidence of edema and reduced blood MMP-9 levels in acute stroke patients [79]. The Glyburide Advantage in Malignant Edema and Stroke (GAMES-RP, clinicaltrials.gov NCT01794182) trial is an ongoing prospective, multicenter randomized double-blind study evaluating glyburide in acute severe anterior circulation stroke patients. The primary outcomes are 90-day functional outcome (mRS ≤4) without decompressive craniectomy .
MMP-9 inhibitors
-
MMP-9 is activated in acute stroke and promotes BBB breakdown and development of edema. High circulating levels after stroke are associated with worse outcomes. The antibiotic minocycline is known to inhibit MMP-9. Two small trials demonstrated safety when used in acute stroke patients and to lower MMP-9 levels [80, 81]. The Minocycline to Improve Neurologic Outcome in Stroke (MINOS, clinicaltrials.gov identifier NCT00630396) open label phase 2 trial has been completed but results have not yet been published.
-
Edaravone is a free radical scavenger that inhibits MMP-9 and has been used as a neuroprotective agent in Japan and China in conjunction with IV thrombolysis. A 2011 Cochrane review [82] found a trend toward effectiveness in published studies, though there is a need for larger high quality trials.
Progesterone
-
Progesterone is proposed to be neuroprotective through effects on BBB and prevention of edema formation. A phase-II trial in traumatic brain injury suggested mortality benefit [83]. There is no data available currently to make recommendations in acute stroke.
Bumetanide
-
As mentioned above, the NKCCl transporter is up-regulated in the setting of ischemic injury and may contribute to edema formation. Bumetanide a small molecule with a well established safety profile , is a relatively specific inhibitor of NKCCl at low concentrations [84]. Trials to evaluate its effect on brain injury including trauma and stroke are currently being designed [18•].
Ethical considerations
Decision making for patients with large ischemic stroke with cerebral edema is a shared process between physicians and families. Level of disability and perceived quality of life may not correlate. That being said, physicians should communicate with families in an open and honest way about prognosis. Mortality in large hemispheric strokes remains between 20 % and 30 % despite best medical and surgical interventions. The vast majority of survivors are markedly disabled with approximately one-third needing continuous nursing care. Families should be prepared for the possibility of depression, disihibition, lack of initiative, irritability, and being wheelchair bound. A significant proportion will likely need tracheostomy and gastrostomy. The outcome of cerebellar hemispheric strokes is often good in the absence of brainstem infarctions, making decision-making less problematic [37].
Conclusions
Malignant edema after ischemic infarctions is associated with significant morbidity and mortality despite current best medical practices including osmotic therapies and decompressive surgeries.
Medical interventions designed to lower global ICP have not been shown to improve outcomes but may delay neurologic decline prior to definitive (surgical) treatment.
While decompressive surgery is associated with good outcomes in hemispheric cerebellar strokes, a survival benefit in large anterior circulation strokes needs to be balanced with the likelihood of marked disability.
Recent advances in understanding the development of edema after ischemia are helping to identify potential targets for interventions to prevent edema development. Of particular interest are the ongoing GAMES-RP and ICTuS 2/3 trials looking at glyburide and hypothermia, respectively. The Neurocritical Care Society will be issuing a guideline soon on the management of large hemispheric infarctions.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Silver FL, Norris JW, Lewis AJ, Hachinski VC. Early mortality following stroke: a prospective review. Stroke. 1984;15:492–6.
Moulin DE, Lo R, Chiang J, Barnett HJ. Prognosis in middle cerebral artery occlusion. Stroke. 1985;16:282–4.
Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R. ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol. 1996;53:309–15.
Simard JM, Sahuquillo J, Sheth KN, Kahle KT, Walcott BP. Managing malignant cerebral infarction. Curr Treat Options Neurol. 2011;13:217–29.
Kaste M, Waltimo O. Prognosis of patients with middle cerebral artery occlusion. Stroke. 1976;7:482–5.
Walcott BP, Miller JC, Kwon CS, et al. Outcomes in severe middle cerebral artery ischemic stroke. Neurocrit Care. 2014;21:20–6.
Manno EM. The management of large hemispheric cerebral infarcts. Compr Ther. 2005;31:124–30.
Huttner HB, Schwab S. Malignant middle cerebral artery infarction: clinical characteristics, treatment strategies, and future perspectives. Lancet Neurol. 2009;8:949–58.
Jaramillo A, Gongora-Rivera F, Labreuche J, Hauw JJ, Amarenco P. Predictors for malignant middle cerebral artery infarctions: a postmortem analysis. Neurology. 2006;66:815–20.
Kimberly WT, Sheth KN. Approach to severe hemispheric stroke. Neurology. 2011;76:S50–6.
Ropper AH. Lateral displacement of the brain and level of consciousness in patients with an acute hemispheral mass. New Engl J Med. 1986;314:953–8.
Cucchiara BL, Kasner SE, Wolk DA, et al. Early impairment in consciousness predicts mortality after hemispheric ischemic stroke. Crit Care Med. 2004;32:241–5.
Blacker DJ, Wijdicks EF. Delayed complete bilateral ptosis associated with massive infarction of the right hemisphere. Mayo Clin Proc. 2003;78:836–9.
Park MH, Kim BJ, Koh SB, Park MK, Park KW, Lee DH. Lesional location of lateral medullary infarction presenting hiccups (singultus). J Neurol Neurosurg Psychiatry. 2005;76:95–8.
Koh MG, Phan TG, Atkinson JL, Wijdicks EF. Neuroimaging in deteriorating patients with cerebellar infarcts and mass effect. Stroke. 2000;31:2062–7.
Hwang DY, Silva GS, Furie KL, Greer DM. Comparative sensitivity of computed tomography vs. magnetic resonance imaging for detecting acute posterior fossa infarct. J Emerg Med. 2012;42:559–65.
Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–68.
Walcott BP, Kahle KT, Simard JM. Novel treatment targets for cerebral edema. Neurotherapeutics. 2012;9:65–72. Outlines recent advances in understanding of the pathophysiology of cerebral edema and potential novel targets for treatment.
Simard JM, Geng Z, Silver FL, et al. Does inhibiting Sur1 complement rt-PA in cerebral ischemia? Ann N Y Acad Sci. 2012;1268:95–107.
Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D. Inhibition of Na(+)-K(+)-Cl(-) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961:22–31.
Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–78.
Hofmeijer J, Algra A, Kappelle LJ, van der Worp HB. Predictors of life-threatening brain edema in middle cerebral artery infarction. Cerebrovasc Dis. 2008;25:176–84.
Maramattom BV, Bahn MM, Wijdicks EF. Which patient fares worse after early deterioration due to swelling from hemispheric stroke? Neurology. 2004;63:2142–5.
Krieger DW, Demchuk AM, Kasner SE, Jauss M, Hantson L. Early clinical and radiological predictors of fatal brain swelling in ischemic stroke. Stroke. 1999;30:287–92.
Kasner SE, Demchuk AM, Berrouschot J, et al. Predictors of fatal brain edema in massive hemispheric ischemic stroke. Stroke. 2001;32:2117–23.
Wang KW, Chang WN, Ho JT, et al. Factors predictive of fatality in massive middle cerebral artery territory infarction and clinical experience of decompressive hemicraniectomy. Eur J Neurol. 2006;13:765–71.
Oppenheim C, Samson Y, Manai R, et al. Prediction of malignant middle cerebral artery infarction by diffusion-weighted imaging. Stroke. 2000;31:2175–81.
Thomalla GJ, Kucinski T, Schoder V, et al. Prediction of malignant middle cerebral artery infarction by early perfusion- and diffusion-weighted magnetic resonance imaging. Stroke. 2003;34:1892–9.
Foerch C, Otto B, Singer OC, et al. Serum S100B predicts a malignant course of infarction in patients with acute middle cerebral artery occlusion. Stroke. 2004;35:2160–4.
Serena J, Blanco M, Castellanos M, et al. The prediction of malignant cerebral infarction by molecular brain barrier disruption markers. Stroke. 2005;36:1921–6.
Manno EM, Nichols DA, Fulgham JR, Wijdicks EF. Computed tomographic determinants of neurologic deterioration in patients with large middle cerebral artery infarctions. Mayo Clin Proc. 2003;78:156–60.
Pullicino PM, Alexandrov AV, Shelton JA, Alexandrova NA, Smurawska LT, Norris JW. Mass effect and death from severe acute stroke. Neurology. 1997;49:1090–5.
Kucinski T, Koch C, Grzyska U, Freitag HJ, Kromer H, Zeumer H. The predictive value of early CT and angiography for fatal hemispheric swelling in acute stroke. AJNR. 1998;19:839–46.
Thomalla G, Hartmann F, Juettler E, et al. Prediction of malignant middle cerebral artery infarction by magnetic resonance imaging within 6 hours of symptom onset: a prospective multicenter observational study. Ann Neurol. 2010;68:435–45.
Haring HP, Dilitz E, Pallua A, et al. Attenuated corticomedullary contrast: an early cerebral computed tomography sign indicating malignant middle cerebral artery infarction. A case-control study. Stroke. 1999;30:1076–82.
Ryoo JW, Na DG, Kim SS, et al. Malignant middle cerebral artery infarction in hyperacute ischemic stroke: evaluation with multiphasic perfusion computed tomography maps. J Comput Assist Tomogr. 2004;28:55–62.
Wijdicks EF, Sheth KN, Carter BS, et al. Recommendations for the management of cerebral and cerebellar infarction with swelling: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:1222–38.
Nagai M, Hoshide S, Kario K. The insular cortex and cardiovascular system: a new insight into the brain-heart axis. J Am Soc Hypertens. 2010;4:174–82.
de Courten-Myers GM, Kleinholz M, Holm P, et al. Hemorrhagic infarct conversion in experimental stroke. Ann Emerg Med. 1992;21:120–6.
Gilmore RM, Stead LG. The role of hyperglycemia in acute ischemic stroke. Neurocrit Care. 2006;5:153–8.
Rosso C, Corvol JC, Pires C, et al. Intensive versus subcutaneous insulin in patients with hyperacute stroke: results from the randomized INSULINFARCT trial. Stroke. 2012;43:2343–9.
Chen H, Chopp M, Welch KM. Effect of mild hyperthermia on the ischemic infarct volume after middle cerebral artery occlusion in the rat. Neurology. 1991;41:1133–5.
Diringer MN, Reaven NL, Funk SE, Uman GC. Elevated body temperature independently contributes to increased length of stay in neurologic intensive care unit patients. Crit Care Med. 2004;32:1489–95.
Fulgham JR, Ingall TJ, Stead LG, Cloft HJ, Wijdicks EF, Flemming KD. Management of acute ischemic stroke. Mayo Clin Proc. 2004;79:1459–69.
Jauch EC, Saver JL, Adams HPJ, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870–947.
Frank JI. Large hemispheric infarction, deterioration, and intracranial pressure. Neurology. 1995;45:1286–90.
Manno EM, Adams RE, Derdeyn CP, Powers WJ, Diringer MN. The effects of mannitol on cerebral edema after large hemispheric cerebral infarct. Neurology. 1999;52:583–7.
Videen TO, Zazulia AR, Manno EM, et al. Mannitol bolus preferentially shrinks non-infarcted brain in patients with ischemic stroke. Neurology. 2001;57:2120–2.
Poca MA, Benejam B, Sahuquillo J, et al. Monitoring intracranial pressure in patients with malignant middle cerebral artery infarction: is it useful? J Neurosurg. 2010;112:648–57.
Raco A, Caroli E, Isidori A, Salvati M. Management of acute cerebellar infarction: one institution’s experience. Neurosurgery. 2003;53:1061–5. discussion 1065–6.
Muizelaar JP, Wei EP, Kontos HA, Becker DP. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg. 1983;59:822–8.
Diringer MN, Scalfani MT, Zazulia AR, Videen TO, Dhar R. Cerebral hemodynamic and metabolic effects of equi-osmolar doses mannitol and 23.4% saline in patients with edema following large ischemic stroke. Neurocrit Care. 2011;14:11–7.
Cruz J, Minoja G, Okuchi K, Facco E. Successful use of the new high-dose mannitol treatment in patients with Glasgow Coma Scale scores of 3 and bilateral abnormal pupillary widening: a randomized trial. J Neurosurg. 2004;100:376–83.
Garcia-Morales EJ, Cariappa R, Parvin CA, Scott MG, Diringer MN. Osmole gap in neurologic-neurosurgical intensive care unit: its normal value, calculation, and relationship with mannitol serum concentrations. Crit Care Med. 2004;32:986–91.
Bereczki D, Fekete I, Prado GF, Liu M. Mannitol for acute stroke. Cochrane Database Syst Rev 2007 Jul 18;(3):CD001153.
Koenig MA, Bryan M, Lewin 3rd JL, Mirski MA, Geocadin RG, Stevens RD. Reversal of transtentorial herniation with hypertonic saline. Neurology. 2008;70:1023–29.
Toung TJ, Hurn PD, Traystman RJ, Bhardwaj A. Global brain water increases after experimental focal cerebral ischemia: effect of hypertonic saline. Crit Care Med. 2002;30:644–9.
Ziai WC, Toung TJ, Bhardwaj A. Hypertonic saline: first-line therapy for cerebral edema? J Neurol Sci. 2007;261:157–66.
Diringer MN, Zazulia AR. Osmotic therapy: fact and fiction. Neurocrit Care. 2004;1:219–33.
Jauss M, Krieger D, Hornig C, Schramm J, Busse O. Surgical and medical management of patients with massive cerebellar infarctions: results of the German-Austrian Cerebellar Infarction Study. J Neurol. 1999;246:257–64.
Vahedi K, Hofmeijer J, Juettler E, et al. Early decompressive surgery in malignant infarction of the middle cerebral artery: a pooled analysis of three randomised controlled trials. Lancet Neurol. 2007;6:215–22.
Vahedi K, Vicaut E, Mateo J, et al. Sequential-design, multicenter, randomized, controlled trial of early decompressive craniectomy in malignant middle cerebral artery infarction (DECIMAL Trial). Stroke. 2007;38:2506–17.
Juttler E, Schwab S, Schmiedek P, et al. Decompressive Surgery for the Treatment of Malignant Infarction of the Middle Cerebral Artery (DESTINY): a randomized, controlled trial. Stroke. 2007;38:2518–25.
Hofmeijer J, Kappelle LJ, Algra A, et al. Surgical decompression for space-occupying cerebral infarction (the Hemicraniectomy After Middle Cerebral Artery infarction with Life-threatening Edema Trial [HAMLET]): a multicentre, open, randomised trial. Lancet Neurol. 2009;8:326–33.
Zhao J, Su YY, Zhang Y, et al. Decompressive hemicraniectomy in malignant middle cerebral artery infarct: a randomized controlled trial enrolling patients up to 80 years old. Neurocrit Care. 2012;17:161–71.
Juttler E, Unterberg A, Woitzik J, et al. Hemicraniectomy in older patients with extensive middle-cerebral-artery stroke. New Engl J Med. 2014;370:1091–100.
Frank JI, Schumm LP, Wroblewski K, et al. Hemicraniectomy and durotomy upon deterioration from infarction-related swelling trial: randomized pilot clinical trial. Stroke. 2014;45:781–7.
Kilincer C, Asil T, Utku U, et al. Factors affecting the outcome of decompressive craniectomy for large hemispheric infarctions: a prospective cohort study. Acta Neurochir. 2005;147:587–94. discussion 594.
Manawadu D, Quateen A, Findlay JM. Hemicraniectomy for massive middle cerebral artery infarction: a review. Can J Neurol Sci. 2008;35:544–50.
Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev. 2011 Sep 7;(9):CD000064.
Smith DS, Rehncrona S, Siesjo BK. Barbiturates as protective agents in brain ischemia and as free radical scavengers in vitro. Acta Physiol Scand Suppl. 1980;492:129–34.
Schwab S, Spranger M, Schwarz S, Hacke W. Barbiturate coma in severe hemispheric stroke: useful or obsolete? Neurology. 1997;48:1608–13.
Dietrich WD. The importance of brain temperature in cerebral injury. J Neurotrauma. 1992;9 Suppl 2:S475–85.
Castillo JMF, Leira R, Prieto JM, Lema M, Noya M. Mortality and morbidity of acute cerebral infarction related to temperature and basal analytic parameters. Cerebrovasc Dis. 1994;4:66–71.
Kammersgaard LP, Jorgensen HS, Rungby JA, et al. Admission body temperature predicts long-term mortality after acute stroke: the Copenhagen Stroke Study. Stroke. 2002;33:1759–62.
Lyden PD, Hemmen TM, Grotta J, Rapp K, Raman R. Endovascular therapeutic hypothermia for acute ischemic stroke: ICTuS 2/3 protocol. Intl J Stroke. 2014;9:117–25.
Kunte H, Schmidt S, Eliasziw M, et al. Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke. Stroke. 2007;38:2526–30.
Kunte H, Busch MA, Trostdorf K, et al. Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann Neurol. 2012;72:799–806.
Kimberly WT, Battey TW, Pham L, et al. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care. 2014;20:193–201.
Fagan SC, Waller JL, Nichols FT, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41:2283–7.
Switzer JA, Hess DC, Ergul A, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke. 2011;42:2633–5.
Feng S, Yang Q, Liu M, et al. Edaravone for acute ischaemic stroke. Cochrane Database Syst Rev. 2011 Dec 7;(12):CD007230.
Wright DW, Kellermann AL, Hertzberg VS, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391–402. 402 e391–392.
Staley KJ. Wrong-way chloride transport: is it a treatable cause of some intractable seizures? Epilepsy Curr. 2006;6:124–7.
Compliance with Ethics Guidelines
Conflict of Interest
Michael E. Brogan and Edward M. Manno declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Cerebrovascular Disorders
Rights and permissions
About this article

Cite this article
Brogan, M.E., Manno, E.M. Treatment of Malignant Brain Edema and Increased Intracranial Pressure After Stroke. Curr Treat Options Neurol 17, 327 (2015). https://doi.org/10.1007/s11940-014-0327-0
Published:
DOI: https://doi.org/10.1007/s11940-014-0327-0