
Abstract
Purpose of the Review
Cryoglobulins are immunoglobulins with the ability to precipitate at temperatures <37 °C. They are related to hematological disorders, infections [especially hepatitis C virus (HCV)], and autoimmune diseases. In this article, the state of the art on Cryoglobulinemic Vasculitis (CV), in a helpful and schematic way, with a special focus on HCV related Mixed Cryoglobulinemia treatment are reviewed.
Recent Findings
Direct – acting antivirals (DAA) against HCV have emerged as an important key in HCV treatment to related Cryoglobulinemic Vasculitis, and should be kept in mind as the initial treatment in non–severe manifestations. On the other hand, a recent consensus panel has published their recommendations for treatment in severe and life threatening manifestations of Mixed Cryoglobulinemias.
Summary
HCV-Cryoglobulinemic vasculitis is the most frequent form of CV. There are new treatment options in HCV-CV with DAA, with an important number of patients achieving complete response and sustained virologic response (SVR). In cases of severe forms of CV, treatment with Rituximab and PLEX are options. The lack of data on maintenance therapy could impulse future studies in this setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction and Definitions
Cryoglobulins are immunoglobulins (Ig) that precipitate in vitro at cold temperature and dissolve at 37 °C, while Cryoglobulinemia (CG) is defined as the presence of cryoglobulins in serum [1, 2]. It must be noted that CG is a laboratory finding and is not necessarily associated with any signs or disease symptoms. Cryoglobulinemic Vasculitis (CV) corresponds to the presence of clinical manifestations due to immune complex deposition along medium and small vessel walls [3]. The presence of symptoms without a positive test for serum cryoglobulins may occur because of an important deposition of the total pool of cryoprecipitable immunocomplexes in targeted vessels and due to false negative results owing to improper blood sampling or inadequate laboratory processes [4]. Due to the latter, cryoglobulin testing should be serially rechecked when CV is highly suspected. On the other hand, healthy people can have CG at low concentrations and infections can trigger the appearance of serum cryoglobulins temporarily without symptoms [3,4,5].
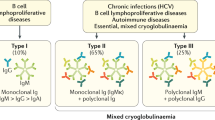
Since 1974, CG has usually been classified in three subgroups according to immunoglobulin composition (“Brouet’s classification”): Type I CG corresponds to one monoclonal isotype or subclass of Ig, typically IgM. It is always associated with hematological disorders like Multiple Myeloma (MM), Waldenstrom’s Macroglobulinemia (WM), Monoclonal Gammopathy of Undetermined Significance (MGUS), Chronic Lymphocytic Leukemia (CLL) and B Cell Non-Hodgkin Lymphoma (B-NHL). Type II and III CGs are typically composed of IgM and IgG, so they are called Mixed Cryoglobulinemias (MC). Characteristically the IgM component of MCs has rheumatoid factor activity so can form immune complexes. Type II CG is usually conformed of a combination of monoclonal IgM and polyclonal IgG while type III CG presents both polyclonal IgM and IgG [3, 6, 7••] (Fig. 1). The presence of oligoclonal IgM or a mixture of monoclonal and polyclonal IgM plus polyclonal IgG have been described as type II – III mixed CG and is considered a transition from type III to type II CG [7••, 8].

Understanding the Cryoglobulinemias. CG: cryoglobulinemia, RF: Rheumatoid factor. Note that type I CGs are almost IgM, less frequently IgG and rarely IgA
Causes
The percentage of each type of cryoglobulin in total number of patients with CG and the most important causes related to CG are described in Table 1.
Epidemiology
CV is a rare disease with an estimated prevalence of less than 5 cases per 100,000 patients in Europe and North America. Information on the prevalence in the rest of the world is scant, but the highest prevalence has been described in the Mediterranean basin [7••].
It is known that 80–90% of MC cases are caused by HCV infection and the presence of CG depends on how long the individual has been infected (annual incidence of 3%) [11,12,13,14]. On the other hand, 40–60% of patients infected by HCV have Mixed Cryoglobulinemia while only 5% of these infected patients will develop CV [3, 11, 15]. Interestingly, for unknown reasons, patients infected with HCV from the Mediterranean basin have the highest prevalence of MC and this group of patients is more prone to be infected with HCV [3]. When comparing patients with HCV chronic infection, with and without Cryoglobulinemia, the former have higher incidence of cirrhosis and higher fibrosis scores [16, 17].
As previously described, other viruses have been related to the presence of MC. Findings of importance are that 7–17% of HIV patients have Cryoglobulins in their serum, while the coinfection of HIV/HCV increases the percentage to 35–64% [3, 18]. About 2% of CV is due to HBV infection [19].
MC has been observed in up to 25% of patients with Systemic Lupus Erythematosus and 10% of patients with Rheumatoid Arthritis [3, 20]. In patients with primary Sjogren’s Syndrome (pSS), 5–20% may have type II CG [21]. The latter association is related with extra glandular involvement, development of systemic vasculitis and B cell lymphoma in pSS patients [22].
Relationship between MC related to HCV and B - NHL
An Italian retrospective multicenter study of 1255 patients with symptomatic MC related to HCV, showed that these patients are 35 times more likely to develop B-NHL than people without MC, especially with low grade lymphomas [23, 24]. Some risk factors for the appearance of B-NHL in these patients were the presence of a symptomatic CG, more than 0.6 g/L of cryoglobulins in serum and low immunoglobulin levels [7••].
Pathophysiology and Disease Mechanisms
The continuum of production of cryoglobulins and the presence of clinical manifestations in Cryoglobulinemia may be summarized in three steps [3, 7••]: (1) In the context of lymphoproliferative disorders or a chronic stimulation of B lymphocytes (e.g. chronic infections or rheumatic diseases) there will be a monoclonal or polyclonal expansion of B cells (BC) in type I/II and III CG, respectively. (2) The subsequent synthesis of mono, oligo or polyclonal cryoglobulins. (3) Tissue injury due to Cryoglobulinemia; with vessel occlusion and hyper viscosity in type I CG and formation/deposit of immune complexes in type II/III CG.
The fact that cryoglobulins are cold insoluble is not fully understood [4]. The diminished presence of tyrosine in the molecule with a relative majority of hydrophobic amino acids; a smaller amount of galactose and sialic acid at Fc region of these immunoglobulins, and large IgM – IgG immune complexes in type II CG have been related to cold precipitation [4, 25, 26]. Because manifestations of CG are not exclusive to acral zones of the body (e.g. kidney compromise), other factors as pH, ionic strength, concentration of cryoglobulins in plasma or different local visceral concentration of chloride or calcium are also implicated in aggregation of cryoglobulins [4, 27,28,29].
HCV infection Model
The most studied model corresponds to type II MC due to HCV infection. The E2 protein from envelope of HCV interacts with CD81, a transmembrane protein that is present in B lymphocytes (working as a complex with CD19 and CD21) and hepatocytes [30, 31]. Of consideration, the viral replication is seen in CD19 peripheral blood mononuclear cells (PBMC) but not in CD19 negative PBMC [32]. Once activated by HCV, the latter complex mentioned between CD81, CD19 and CD21 triggers a polyclonal expansion that apparently is not related to B cell receptor (BCR) stimulation [7••, 33]; also, an upregulation of B cell activator factor (BAFF/Blys) has been described [34, 35]. Importantly, the hallmark of type II MC is the presence of B cell clones that produce monoclonal IgM (the WA cross-reacting idiotype) with rheumatoid factor activity against polyclonal IgG, which in turn binds to HCV core protein [3, 7••, 36,37,38]. These B cell clones are present as lymphoid infiltrates in the liver and other sites as bone marrow, spleen and peripheral blood [3, 7••]. Of note, beside cryoglobulins, HCV infection stimulates the production of various other antibodies that determine the presence of other autoimmune manifestations in the context of a “HCV syndrome” [39] (Table 2).
The immune dysregulation characterized by HCV infection and the presence of CG have been described as risk factors for lymphomagenesis [40,41,42]. In addition to chronic stimulation of B cells, other mechanisms related to HCV infection and the transformation from polyclonal to monoclonal BC are: (a) The presence of mutations in genes related to heavy and light immunoglobulin (Ig) chains [43]; (b) Overexpression of BCL-2 oncogene due to augmented t(14:18) translocation and the overexpression of other oncogenes as MYC [7••, 44]; (c) The role of certain viral proteins (NS3 and core proteins) that cause alterations in DNA repair and (d) activation of different intracellular signaling pathways as JNK and ERK or transcription factors as NF-kB [42].
As discussed above, tissue injury in type I CG is derived from vessel occlusion with high concentrations of CGs. On the other hand, immune – complex (IC) deposition and subsequent complement activation in MC due to HCV infection is the most important mechanism. The ICs containing HCV core proteins also contain C1q protein which can bind to the receptor for the globular domain of C1q protein (gC1q-R) in the membrane of endothelial cells, a process that can be related to deposition and posterior vascular damage [38, 45]. Of note, the latter ICs that bind to gC1qR can promote activation of complement system (classical pathway) and deposition of C4, a fact that could explain the characteristically low C4 in these patients [46••]. Moreover, CGs escape from the erythrocyte transport system and the HCV infection interferes with the lysosomal activity of phagocytes so there is a diminished blood clearance of these ICs [47, 48].
Clinical Presentation
Type I CG
The typical clinical picture of type I CG is related to vessel occlusion due to a characteristic ability for cryoprecipitation, but some vasculitis features can be seen too [9]. In this context, the presence of cold – induced symptoms (90–100%), purpura (80%), Raynaud phenomenon (25–40%) that sometimes can be complicated with digital gangrene; distal ulcers with necrosis (30–35%) and peripheral neuropathy (30–50%) are important findings. Less frequently, renal compromise, arthralgias and livedo reticularis are seen [49, 50]. Cryocrit is usually above 5% and can be higher than 50%. In the case of high cryocrit and therefore a high serum viscosity (> than 4.0 cP habitually), hyperviscosity syndrome (HS) could develop. Some features of HS are mental confusion and headache; visual loss or blurred vision; nasal bleeding and hypoacusia [3, 9].
Type II & III CG (MC)
Due to RF activity and consequent immune complexes formation and activation of the complement system, the clinical manifestations of MC are related to small and medium vessel vasculitis [3, 9]. CV is characterized by a triad of purpura, weakness and arthralgias (“Meltzer’s triad”). Also, a variable combination of symptoms can be associated, including: skin ulcers, diffuse vasculitis, peripheral neuropathy, membranoproliferative glomerulonephritis (MPGN), chronic hepatitis, lymphatic and hepatic malignancies. Classic laboratory findings are low complement levels (specially C4) and positive RF. Cryocrit is lower than 5% in MC [9].
-
Cutaneous: The most common manifestation of MC is purpura (90–98%) of the lower limbs which might be orthostatic and intermittent and is a key clue for diagnosis, often complicated with ulcers or hyperpigmentation at resolution [19]. Cutaneous ulcers and livedo reticularis traduce compromise of medium - sized vessels [3]. The typical histopathological pattern of CV is the presence of non-necrotizing leukocytoclastic vasculitis of small and medium vessels. Necrotizing vasculitis with fibrinoid necrosis can be seen in 20% [10].
-
Articular: Frequently arthralgias (44–71%). Less often, a non-erosive oligoarthritis can be seen(10%) [3] .
-
Sicca syndrome (50%) [39].
-
Neurologic: Neuropathies occur in 65% of HCV - related MC by clinical criteria and 80% by electrodiagnostic testing (ET); polyneuropathies are more common than mononeuritis multiplex [2, 51,52,53]. Most neuropathies are distal, slowly progressive, and sensorimotor (but sensory predominant). About 50% of neuropathies are painful, and approximately 25% of them are small fiber in character [54]. ET studies reveal an axonal, sensorimotor neuropathy and nerve biopsies reveal predominantly axonal alterations with mononuclear predominant perivascular inflammation in the epineurium, with definite vasculitis in 50% [55].
Central nervous system (CNS) involvement is rarely seen but may appear with dysarthria and hemiplegia [2].
-
Renal: Renal involvement (RI) has been reported in 18–40% of patients with CV and these patients have higher mortality [19, 56,57,58]. The occurrence of RI follows the diagnosis of Cryoglobulinemia after 2.6–4 years and occurs until 41 years after diagnosis [59, 60]. It is mainly characterized by urinary abnormalities: proteinuria is seen in 88–100% of cases and hematuria in almost all patients. Elevation of plasma creatinine is described in 47–63% of patients with RI [59, 60]. Kidney biopsy must be performed for confirmation of RI in CV: the histopathological features observed are characterized by a glomerular monocyte infiltration with double contours of the basement membrane and hyaline intraluminal thrombi, similar to a type I membranoproliferative glomerulonephritis (MPGN) [61]. Immunofluorescence analysis defines the RI as an immune complex-mediated MPGN, showing intra-glomerular sub-endothelial deposits of Ig identical to those of the cryoprecipitate, and complement components. Renal necrotizing vasculitis and extra- capillary crescents are rarely observed [61, 62]. Some predictive factors of RI in CV are the presence of Type II CG, a high cryocrit, an IgG kappa monoclonal component and diabetes [63].
-
Liver / Gastrointestinal (GI): Due to HCV infection, over 70% of patients develop chronic hepatitis. This has a mild to moderate clinical course, but may evolve to cirrhosis in 25% of patients. Around 3% of patients develop hepatocellular carcinoma [39]. GI tract compromise has been observed in 2–6% of patients. Some manifestations are abdominal pain, GI bleeding and perforation due to intestinal ischemia [3, 64].
-
Hyper viscosity Syndrome: Rare manifestation, < 3% in MC [65].
-
Respiratory system (RS): The compromise of the RS accounts for less than 5%. The presence of interstitial lung disease, alveolar hemorrhage due to vasculitis and pleural effusion have been described [3].
-
Diffuse vasculitis: Severe and unusual complication of the disease. Involves medium to small vessels with multiple organ involvement (skin, lungs, kidneys, gastrointestinal and CNS) [2].
-
Endocrinological disorders: More frequent than in patients without CV. Thyroid disorders (including cancer); diabetes mellitus type 2 and erectile dysfunction can be seen [66,67,68].
-
Cardiovascular disease: Reports of myocarditis, myocardial infarction, heart failure and pericarditis have been published [69, 70].
Some manifestations are considered as life – threatening (LT) events (GI ischemia, RI, alveolar hemorrhage and CNS compromise) (Table 3). In a Spanish series of 209 patients diagnosed with CG between 1991 and 1999, 14% of them developed LT manifestations of CG. Of these, 59% presented LT events as the initial manifestation of the disease and 66% died. Interestingly, intestinal ischemia and pulmonary hemorrhage had 100% of mortality. With regard to etiology, 76% had associated chronic viral infection (HCV and HIV) and 31% of patients had autoimmune diseases. Patients with LT Cryoglobulinemic Vasculitis had a higher frequency of fever, type II cryoglobulins, low C3 levels and a higher mean value of cryocrit than patients with milder forms of CV [69, 71].
Diagnosis
Diagnosis is based on history, clinical manifestations of the disease and the presence of cryoglobulins in serum. It is necessary to rule out differential diagnosis as Rheumatoid Arthritis, primary Sjögren’s syndrome, B cell neoplasms and MPGN. Also, the viral status of the patient must be explored (HCV, HBV, HIV) [7••, 39].
For the detection of cryoglobulins, 10 to 20 mL of blood is drawn into collection tubes, prewarmed to 37 °C without anticoagulants. After clotting at 37 °C for 30 to 60 min, the serum is separated by centrifugation at 37 °C, placed in a graduated (Wintrobe) tube and refrigerated (4 °C) to allow the precipitation of cryoglobulin for 3–7 days. After that, if positive, a precipitate should appear at the bottom of the tube. If the precipitate is truly cryoglobulins, it will re-dissolve when the tube is heated at 37 °C. In this process, pre-analytical conditions are probably the most important. If the temperature of the pre-analytical conditions is not controlled to prevent cooling, cryoglobulins may already precipitate during this stage and give false negative results [72].
The next step for laboratories is to determine a cryocrit, which is a measure of the packed (centrifuged) volume of the precipitate as a percentage of the original serum volume at 4 °C. The cryocrit in individuals without CG is close to zero and a cryocrit over 0.5 to 1% or cryoglobulin concentration above 2 to 5 mg/dL is considered clinically significant [59]. It should be kept in mind that there is no strict relationship between the cryoglobulin concentration and the severity of the clinical manifestations [56]. Posteriorly, the precipitate can be used for typing and quantification of the cryoprecipitate [72]. Typing of the cryoglobulin is normally performed by immunofixation or immune-electrophoresis. Typing enables identification of the cryoprecipitate as immunoglobulin, and allows to make clinical associations and prevent possible complications for the three types of cryoglobulins [73].
Treatment
The treatment of cryoglobulinemic manifestations depends on the type of CG, the etiology and its severity. It should be noted that cold and long standing avoidance must be encouraged in all patients [9].
In type I CG the basic principle of treatment will be the management of the underlying hematologic disorder [9, 10]. As discussed before, CLL, MM, WM, MGUS or B-NHL are the most common malignancies associated with type I CG [7••, 9] . The use of steroids plus Rituximab (RTX), Bortezomib, Lenalidomide or other agents as Cyclophosphamide have been described as therapeutic options in a large American series of 102 patients with type I CG and WM guidelines [49, 74]. In the case of hyper viscosity symptoms, plasma exchange (PLEX) should be performed together with the agents exposed above [9]. Of note, it must be considered that RTX has been associated with hyper viscosity syndrome due to IgM flare in WM patients, so preemptive plasmapheresis before RTX should be considered in the case of a basal IgM of >4 g/dl [9, 75].
The treatment of Cryoglobulinemic vasculitis (CV) in the context of MC must be divided into infectious or non- infectious causes. The next step is to classify CV manifestations as non-severe, severe or life threatening (Table 3) [9]. In the case of infectious CV due to HCV, non-severe symptoms can be treated with direct antiviral agents (DAA) alone. DAA has emerged as a “game changer” in the treatment of HCV virus, with a sustained viral response (SVR) at 12 weeks of 97–100% and less toxic effects than interferon- based schemes. At long term follow up, SVR is associated with a negative detection of HCV RNA and morbimortality associated with HCV [76, 77]. The larger study of CV due to HCV infection is an international, prospective and open - label trial (VASCUVALDIC 3) of 148 patients with HCV associated CV, all of them received a free interferon regimen with DAA for 12 or 24 weeks. At 12 weeks after DAA were stopped, 72% of patients had a complete clinical response (defined as improvement of every organ affected at baseline without a clinical relapse), 22.6% had a partial response and 97.2% had a sustained virologic response (SVR). Cryoglobulinemia disappeared in 53% of patients. Interestingly, a total of 29.1% of patients had a severe form of vasculitis and 14.3% required Steroids, Rituximab or PLEX. Baseline factors independently associated with poor response were peripheral neuropathy and a severe form of CV [78••].
Severe manifestations require the use of glucocorticoids (GC) and immunosuppressants (IS), and DAA can be considered once the patient is stable. Initial use of high dose steroids is needed to quickly subjugate vasculitic damage [78••]. In light of evidence, Rituximab is preferred instead of other immunosuppressive agents in this setting (Table 4) [51, 79, 80•, 81]. Moreover, it seems that RTX at a dose of 250 mg/m2 given two times separated by one week is equally effective as 325 mg/ m2 per week for 4 weeks in CV treatment, and the use of repeated low doses is also a safe and efficacious strategy in case of relapsing disease [82, 83].
PLEX is recommended in life threatening CV (also with CS and IS) in a recent published consensus, on the basis of retrospective studies and only one small randomized trial, that used immunoadsorption apheresis plus immunosuppressive therapy versus the latter alone (Table 4) [69, 80•, 84, 85•]. It should be noted that RTX must be administered after PLEX because the latter removes large quantities of the drug (about half of the dose if PLEX is applied in the first three days after infusion) [86]. Hepatitis B virus (HBV) is a relevant non HCV infectious cause, with frequent relapsing episodes [87]. Albeit the principles of treatment for HBV are the same as HCV infection, antivirals always should be used as first line therapy besides the use of IS and CS in severe and life threatening cases. The previous recommendation is based on the potential risk of flare or reactivation of HBV and HIV with RTX [7••, 9, 88, 89]. Indeed, the use of RTX in active hepatitis flare has been associated with fatal outcomes [90].
In the case of non-infectious CV, the aim of treatment for non–severe manifestations is the management of the underlying disease. When CV is due to essential MC (without apparent cause) low dose CS could be an option [9]. Severe manifestations require the use of IS, specially RTX instead of cyclophosphamide (CYC) according to a large retrospective study of 242 patients from the French multicenter CryoVas survey that showed a better response (but also a greater number of severe infections and similar death rates) with the combination of RTX plus CS versus CYC plus CS or CS alone [91]. The use of PLEX is also encouraged in severe or life threatening cases with warming of the exchange solution [92].
Prognosis
Overall, the survival rates at 10 years are similar for HCV or non-infectious related CV (75% and 79%, respectively). The leading cause of death in CV corresponds to infections (nearly 50% in non-infectious CV and 35% in HCV CV) while other described causes are liver disease (HCV CV), cardiovascular manifestations and organ damage because of vasculitis [58, 93, 94]. The prognosis of patients with symptomatic Cryoglobulinemia is diverse and depends on the organ or system compromised and the underlying disease that caused it. In general, presence of an hematologic neoplasm is an important adverse prognostic factor [10]. The main prognostic baseline factors in HCV related CV are severe liver fibrosis, a high Five-factors score and involvement of heart, kidneys and central nervous system; in non-infectious CV, prognostic factors are: age > 65 years and involvement of lungs, kidneys or gastrointestinal tract [58, 93]. Of interest is that renal compromise could be worst in related HCV than in essential CG [62].
Conclusions
Cryoglobulinemia is a rare disease that acts like a syndrome even within its spectrum of different diseases and in many cases requires a different approach and treatment. Its real prevalence is undetermined mostly due to difficulties in the laboratory processes needed for diagnosis.
However, there are new treatment options for HCV-CV with DAA, with a significant number of patients achieving complete response and SVR; but Rituximab and PLEX are still prescribed in cases that develop severe forms of Vasculitis. The lack of data on maintenance therapy could impulse future studies in this context.
In the setting of Essential Cryoglobulinemia, there is an absence of new treatment options and more research on this subgroup of patients is needed.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ferri C, Zignego AL, Pileri SA. Cryoglobulins. J Clin Pathol [Internet]. 2002;55(1):4–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11825916.
Ferri C. Mixed cryoglobulinemia. Orphanet Journal of Rare Diseases. 2008;3:1–17.
Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet (London, England) [Internet]. 2012;379(9813):348–60 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21868085.
Sargur R, White P, Egner W. Cryoglobulin evaluation: best practice? Ann Clin Biochem [Internet]. 2010;47(Pt 1):8–16 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20040797.
Maire MA, Mittey M, Lambert PH. The presence of cryoprecipitable immunoglobulins in normal human sera may reflect specific molecular interactions. Autoimmunity [Internet]. 1989;2(2):155–64 Available from: http://www.ncbi.nlm.nih.gov/pubmed/2491599.
Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M. Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med. 1974;57(5):775–88.
•• Roccatello D, Saadoun D, Ramos-Casals M, Tzioufas AG, Fervenza FC, Cacoub P, et al. Cryoglobulinaemia. Nat Rev Dis Prim [Internet]. 2018;4(1):11 Available from: http://www.nature.com/articles/s41572-018-0009-4. An excellent, extensive and recent review of cryoglobulinemias.
Tissot JD, Schifferli JA, Hochstrasser DF, Pasquali C, Spertini F, Clément F, et al. Two-dimensional polyacrylamide gel electrophoresis analysis of cryoglobulins and identification of an IgM-associated peptide. J Immunol Methods. 1994;173(1):63–75.
Muchtar E, Magen H, Gertz MA. How I Treat cryoglobulinemia. Blood [Internet]. 2017;129(3):289–98 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27799164.
Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: An update in 2019. Joint Bone Spine [Internet]. 2019 Feb 4; Available from: http://www.ncbi.nlm.nih.gov/pubmed/30731128.
Lunel F, Musset L, Cacoub P, Frangeul L, Cresta P, Perrin M, et al. Cryoglobulinemia in chronic liver diseases: role of hepatitis C virus and liver damage. Gastroenterology. 1994;106:1291–300.
Ferri C, Greco F, Longombardo G, Palla P, Moretti A, Marzo E, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. 1991;34:1606–10.
Cacoub P, Poynard T, Ghillani P, Charlotte F, Olivi M, Charles Piette J, et al. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC group. Multidepartment virus C. Arthritis Rheum. 1999;42(9):2204–12.
Ramos-Casals M, Trejo O, García-Carrasco M, Cervera R, Font J. Mixed cryoglobulinemia: new concepts. Lupus. 2000;9:83–91.
Adinolfi LE, Utili R, Attanasio V, Zampino R, Ragone E, Tripodi MF, et al. Epidemiology, clinical spectrum and prognostic value of mixed cryoglobulinaemia in hepatitis C virus patients: a prospective study. Ital J Gastroenterol. 1996;28:1–9.
Kayali Z, Buckwold VE, Zimmerman B, Schmidt WN. Hepatitis C, cryoglobulinemia, and cirrhosis: a meta- analysis. Hepatology. 2002;36:978–85.
Saadoun D, Asselah T, Resche-Rigon M, Charlotte F, Bedossa P, Valla D, et al. Cryoglobulinemia is associated with steatosis and fibrosis in chronic hepatitis C. Hepatology. 2006;43:1337–45.
Bonnet F, Pineau J-J, Taupin J-L, Feyler A, Bonarek M, de Witte S, et al. Prevalence of cryoglobulinemia and serological markers of autoimmunity in human immunodeficiency virus infected individuals: a cross-sectional study of 97 patients. J Rheumatol. 2003;30(9):2005–10.
Ferri C, Sebastiani M, Giuggioli D, Cazzato M, Longombardo G, Antonelli A, et al. Mixed cryoglobulinemia: demographic, clinical, and serologic features and survival in 231 patients. Semin Arthritis Rheum. 2004;33(6):355–74.
García-Carrasco M, et al. Cryoglobulinemia in systemic lupus erythematosus: prevalence and clinical characteristics in a series of 122 patients. Semin Arthritis Rheum 3. 2001;30:366–73.
Tzioufas AG, Manoussakis MN, Costello R, Silis M, Papadopoulos NM, Moutsopoulos HM. Cryoglobulinemia in autoimmune rheumatic diseases. Evidence of circulating monoclonal cryoglobulins in patients with primary Sjögren’s syndrome. Arthritis Rheum. 1986;29:1098–104.
Brito-Zerón P, et al. Sjögren syndrome. Nat Rev Dis Prim. 2016;(2):16047.
Saadoun D, Landau DA, Calabrese LH, Cacoub PP. Hepatitis C-associated mixed cryoglobulinaemia: a crossroad between autoimmunity and lymphoproliferation. Rheumatology (Oxford) [Internet]. 2007;46(8):1234–42 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17566058.
Monti G, Pioltelli P, Saccardo F, Campanini M, Candela M, Cavallero G, et al. Incidence and characteristics of non-Hodgkin lymphomas in a multicenter case file of patients with hepatitis C virus-related symptomatic mixed cryoglobulinemias. Arch Intern Med [Internet]. 2005;165(1):101–5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/15642884.
Andersen BR, Tesar JT, Schmid FR, Haisty WK, Hartz WH. Biological and physical properties of a human m-cryoglobulin and its monomer subunit. Clin Exp Immunol [Internet]. 1971;9(6):795–807 Available from: http://www.ncbi.nlm.nih.gov/pubmed/5003445.
Trendelenburg M, Schifferli JA. Cryoglobulins in chronic hepatitis C virus infection. Clin Exp Immunol [Internet]. 2003;133(2):153–5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/12869018.
Grey HM, Kohler PF. Cryoimmunoglobulins. Semin Hematol [Internet]. 1973;10(2):87–112 Available from: http://www.ncbi.nlm.nih.gov/pubmed/4633223.
Di Stasio E, Bizzarri P, Casato M, Galtieri A, Fiorilli M, Pucillo LP. Cl- regulates cryoglobulin structure: a new hypothesis for the physiopathological mechanism of temperature non-dependent cryoprecipitation. Clin Chem Lab Med [Internet]. 2004;42(6):614–20 Available from: http://www.ncbi.nlm.nih.gov/pubmed/15259377.
Qi M, Steiger G, Schifferli JA. A calcium-dependent cryoglobulin IgM kappa/polyclonal IgG. J Immunol [Internet]. 1992;149(7):2345–51 Available from: http://www.ncbi.nlm.nih.gov/pubmed/1527381.
Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, et al. Binding of hepatitis C virus to CD81. Science [Internet]. 1998;282(5390):938–41 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9794763.
Charles ED, Dustin LB. Hepatitis C virus-induced cryoglobulinemia. Kidney Int [Internet]. 2009;76(8):818–24 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19606079.
Ito M, Murakami K, Suzuki T, Mochida K, Suzuki M, Ikebuchi K, et al. Enhanced expression of lymphomagenesis-related genes in peripheral blood B cells of chronic hepatitis C patients. Clin Immunol [Internet]. 2010;135(3):459–65 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1521661610000434.
Charles ED, Green RM, Marukian S, Talal AH, Lake-Bakaar GV, Jacobson IM, et al. Clonal expansion of immunoglobulin M+CD27+ B cells in HCV-associated mixed cryoglobulinemia. Blood [Internet]. 2008;111(3):1344–56 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17942751.
Fabris M, Quartuccio L, Sacco S, De Marchi G, Pozzato G, Mazzaro C, et al. B-Lymphocyte stimulator (BLyS) up-regulation in mixed cryoglobulinaemia syndrome and hepatitis-C virus infection. Rheumatology (Oxford) [Internet]. 2007;46(1):37–43 Available from: http://www.ncbi.nlm.nih.gov/pubmed/16735452.
Lake-Bakaar G, Jacobson I, Talal A. B cell activating factor (BAFF) in the natural history of chronic hepatitis C virus liver disease and mixed cryoglobulinaemia. Clin Exp Immunol [Internet]. 2012;170(2):231–7 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23039894.
De Vita S, De Re V, Gasparotto D, Ballarè M, Pivetta B, Ferraccioli G, et al. Oligoclonal non-neoplastic B cell expansion is the key feature of type II mixed cryoglobulinemia: clinical and molecular findings do not support a bone marrow pathologic diagnosis of indolent B cell lymphoma. Arthritis Rheum [Internet]. 2000;43(1):94–102 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10643704.
Dammacco F, Lauletta G, Russi S, Leone P, Tucci M, Manno C, et al. Clinical practice: hepatitis C virus infection, cryoglobulinemia and cryoglobulinemic vasculitis. Clin Exp Med [Internet]. 2019;19(1):1–21 Available from: http://www.ncbi.nlm.nih.gov/pubmed/30430284.
Lauletta G, Russi S, Conteduca V, Sansonno L. Hepatitis C virus infection and mixed cryoglobulinemia. Clin Dev Immunol [Internet]. 2012;2012:502156 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22844322.
Ferri C, Sebastiani M, Giuggioli D, Colaci M, Fallahi P, Piluso A, et al. Hepatitis C virus syndrome: A constellation of organ- and non-organ specific autoimmune disorders, B-cell non-Hodgkin’s lymphoma, and cancer. World J Hepatol [Internet]. 2015;7(3):327–43 Available from: http://www.wjgnet.com/1948-5182/full/v7/i3/327.htm.
Lauletta G, Russi S, Conteduca V, Sansonno L, Dammacco F, Sansonno D. Impact of Cryoglobulinemic Syndrome on the Outcome of Chronic Hepatitis C Virus Infection: A 15-Year Prospective Study. Medicine (Baltimore) [Internet]. 2013;92(5):245–56 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23982056.
Giordano TP, Henderson L, Landgren O, Chiao EY, Kramer JR, El-Serag H, et al. Risk of non-Hodgkin lymphoma and lymphoproliferative precursor diseases in US veterans with hepatitis C virus. JAMA [Internet]. 2007;297(18):2010–7 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17488966.
Carloni G, Fioretti D, Rinaldi M, Ponzetto A. Heterogeneity and coexistence of oncogenic mechanisms involved in HCV-associated B-cell lymphomas. Crit Rev Oncol Hematol [Internet]. 2019;138:156–71 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31092372.
De Re V, De Vita S, Marzotto A, Gloghini A, Pivetta B, Gasparotto D, et al. Pre-malignant and malignant lymphoproliferations in an HCV-infected type II mixed cryoglobulinemic patient are sequential phases of an antigen-driven pathological process. Int J cancer [Internet]. 2000;87(2):211–6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10861476.
Zignego A-L, Giannini C, Ferri C. Hepatitis C virus-related lymphoproliferative disorders: an overview. World J Gastroenterol [Internet]. 2007;13(17):2467–78 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17552031.
Sansonno D, Tucci FA, Ghebrehiwet B, Lauletta G, Peerschke EIB, Conteduca V, et al. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J Immunol [Internet]. 2009;183(9):6013–20 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19828637.
•• El-Shamy A, Branch AD, Schiano TD, Gorevic PD. The Complement System and C1q in Chronic Hepatitis C Virus Infection and Mixed Cryoglobulinemia. Front Immunol [Internet]. 2018;9:1001 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29910796. An interesting review about the important role of C1q and gC1qR in mixed cryoglobulinemia related to HCV infection.
Roccatello D, Isidoro C, Mazzucco G, Mesiti A, Quattrocchio G, Amore A, et al. Role of monocytes in cryoglobulinemia-associated nephritis. Kidney Int [Internet]. 1993;43(5):1150–5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/8510395.
Roccatello D, Morsica G, Picciotto G, Cesano G, Ropolo R, Bernardi MT, et al. Impaired hepatosplenic elimination of circulating cryoglobulins in patients with essential mixed cryoglobulinaemia and hepatitis C virus (HCV) infection. Clin Exp Immunol [Internet]. 1997;110(1):9–14 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9353142.
Sidana S, Rajkumar SV, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol [Internet]. 2017;92(7):668–73 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28370486.
Terrier B, Karras A, Kahn J-E, Le Guenno G, Marie I, Benarous L, et al. The spectrum of type I cryoglobulinemia vasculitis: new insights based on 64 cases. Medicine (Baltimore) [Internet]. 2013;92(2):61–8 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23429354.
De Vita S, Quartuccio L, Isola M, Mazzaro C, Scaini P, Lenzi M, et al. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheum [Internet]. 2012;64(3):843–53 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22147661.
Zaltron S, Puoti M, Liberini P, Antonini L, Quinzanini M, Manni M, et al. High prevalence of peripheral neuropathy in hepatitis C virus infected patients with symptomatic and asymptomatic cryoglobulinaemia. Ital J Gastroenterol Hepatol [Internet]. 1998;30(4):391–5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9789135.
Ciompi ML, Marini D, Siciliano G, Melchiorre D, Bazzichi L, Sartucci F, et al. Cryoglobulinemic peripheral neuropathy: neurophysiologic evaluation in twenty-two patients. Biomed Pharmacother [Internet]. 1996;50(8):329–36 Available from: http://www.ncbi.nlm.nih.gov/pubmed/8952851.
Gemignani F, Brindani F, Alfieri S, Giuberti T, Allegri I, Ferrari C, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry. 2005;76(10):1410–4.
Collins MP. The vasculitic neuropathies. Curr Opin Neurol. 2012;25(5):573–85.
Shihabi ZK. Cryoglobulins: An important but neglected clinical test. Ann Clin Lab Sci. 2006;36:395–408.
Saadoun D, Terrier B, Semoun O, Sene D, Maisonobe T, Musset L, et al. Hepatitis C virus associated polyarteritis nodosa. Arthritis Care Res. 2011;63(3):427–35.
Terrier B, Carrat F, Krastinova E, Marie I, Launay D, Lacraz A, et al. Prognostic factors of survival in patients with non-infectious mixed cryoglobulinaemia vasculitis: data from 242 cases included in the CryoVas survey. Ann Rheum Dis [Internet]. 2013;72(3):374–80 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22586172.
P.D. G, H.J. K, Y. L, R. K, M. M, P. P, et al. Mixed cryoglobulinemia: Clinical aspects and long-term follow-up of 40 patients. Am J Med 1980;69(2):287–308.
Tarantino A, Campise M, Banfi G, Confalonieri R, Bucci A, Montoli A, et al. Long-term predictors of survival in essential mixed cryoglobulinemic glomerulonephritis. Kidney Int. 1995;47(2):618–23.
Roccatello D, Fornasieri A, Giachino O, Rossi D, Beltrame A, Banfi G, et al. Multicenter study on hepatitis C virus-related cryoglobulinemic glomerulonephritis. Am J Kidney Dis [Internet]. 2007;49(1):69–82 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17185147.
Beddhu S, Bastacky S, Johnson JP. The clinical and morphologic spectrum of renal cryoglobulinemia. Medicine (Baltimore) [Internet]. 2002;81(5):398–409 Available from: http://www.ncbi.nlm.nih.gov/pubmed/12352634.
Coliche V, Sarda M-N, Laville M, Chapurlat R, Rheims S, Sève P, et al. Predictive factors of renal involvement in cryoglobulinaemia: a retrospective study of 153 patients. Clin Kidney J [Internet]. 2018;12(3):365–72 Available from: https://academic.oup.com/ckj/article/12/3/365/5168442.
Terrier B, Saadoun D, Sène D, Scerra S, Musset L, Cacoub P. Presentation and outcome of gastrointestinal involvement in hepatitis C virus-related systemic vasculitis: a case-control study from a single-centre cohort of 163 patients. Gut [Internet]. 2010;59(12):1709–15 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20841367.
Della Rossa A, Tavoni A, D’Ascanio A, Catarsi E, Marchi F, Bencivelli W, et al. Mortality rate and outcome factors in mixed cryoglobulinaemia: the impact of hepatitis C virus. Scand J Rheumatol [Internet]. 2010;39(2):167–70 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20337547.
Antonelli A, Ferri C, Fallahi P, Giuggioli D, Nesti C, Longombardo G, et al. Thyroid involvement in patients with overt HCV-related mixed cryoglobulinaemia. QJM [Internet]. 2004;97(8):499–506 Available from: http://www.ncbi.nlm.nih.gov/pubmed/15256607.
Antonelli A, Ferri C, Fallahi P, Sebastiani M, Nesti C, Barani L, et al. Type 2 diabetes in hepatitis C-related mixed cryoglobulinaemia patients. Rheumatology (Oxford) [Internet]. 2004;43(2):238–40 Available from: http://www.ncbi.nlm.nih.gov/pubmed/13130149.
Ferri C, Bertozzi MA, Zignego AL. Erectile dysfunction and hepatitis C virus infection. JAMA [Internet]. 2002;288(6):698–9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/12169072.
Retamozo S, Díaz-Lagares C, Bosch X, Bové A, Brito-Zerón P, Gómez M-E, et al. Life-Threatening Cryoglobulinemic Patients With Hepatitis C: Clinical Description and Outcome of 279 Patients. Medicine (Baltimore) [Internet]. 2013;92(5):273–84 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23974248.
Ali MA, Kayani WZ, Linzie BM, Punjabi G V, Wetmore JB. Myopericarditis in a patient with hepatitis C and cryoglobulinemic renal disease. Clin Case Reports [Internet]. 2017;5(5):616–620. Available from: https://doi.org/10.1002/ccr3.788
Ramos-Casals M, Robles A, Brito-Zerón P, Nardi N, Nicolás JM, Forns X, et al. Life-threatening Cryoglobulinemia: Clinical and immunological characterization of 29 cases. Semin Arthritis Rheum. 2006;36:189–96.
Damoiseaux J, Cohen Tervaert JW. Diagnostics and Treatment of Cryoglobulinaemia: It Takes Two to Tango. Clin Rev Allerg Immu. 2014;47:299–310.
Sargur R, Egner W. Appropriate cryoglobulin investigations - The author responds. Ann Clin Biochem. 2010;47:491–2.
Leblond V, Kastritis E, Advani R, Ansell SM, Buske C, Castillo JJ, et al. Treatment recommendations from the eighth international workshop on Waldenström’s Macroglobulinemia. Blood [Internet]. 2016;128(10):1321–8 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27432877.
Mazzucchelli M, Frustaci AM, Deodato M, Cairoli R, Tedeschi A. Waldenstrom’s Macroglobulinemia: an update. Mediterr J Hematol infect Dis [Internet]. 2018;10(1):e2018004 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29326801.
Simmons B, Saleem J, Hill A, Riley RD, Cooke GS. Risk of Late Relapse or Reinfection With Hepatitis C Virus After Achieving a Sustained Virological Response: A Systematic Review and Meta-analysis. Clin Infect Dis [Internet]. 2016;62(6):683–94 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26787172.
Banerjee D, Reddy KR. Review article: safety and tolerability of direct-acting anti-viral agents in the new era of hepatitis C therapy. Aliment Pharmacol Ther [Internet]. 2016;43(6):674–96 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26787287.
•• Cacoub P, Si Ahmed SN, Ferfar Y, Pol S, Thabut D, Hezode C, et al. Long-term Efficacy of Interferon-Free Antiviral Treatment Regimens in Patients With Hepatitis C Virus-Associated Cryoglobulinemia Vasculitis. Clin Gastroenterol Hepatol [Internet]. 2019;17(3):518–26 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29857143. A recent international and prospective study that confirmed the effectiveness and good tolerance of direct - acting antiviral agents in HCV related cryoglobulinemic vasculitis.
Sneller MC, Hu Z, Langford CA. A randomized controlled trial of rituximab following failure of antiviral therapy for hepatitis C virus-associated cryoglobulinemic vasculitis. Arthritis Rheum [Internet]. 2012;64(3):835–42 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22147444.
• Galli M, Monti G, Marson P, Scaini P, Pietrogrande M, Candela M, et al. Recommendations for managing the manifestations of severe and life-threatening mixed cryoglobulinemia syndrome. Autoimmun rev [Internet]. 2019 Jun; available from: https://linkinghub.elsevier.com/retrieve/pii/S1568997219301351. An evidence based guideline made by an italian consensus panel for treatment of severe and life threatening manifestations of mixed cryoglobulinemias.
Dammacco F, Tucci FA, Lauletta G, Gatti P, De Re V, Conteduca V, et al. Pegylated interferon-α, ribavirin, and rituximab combined therapy of hepatitis C virus-related mixed cryoglobulinemia: A long-term study. Blood. 2010;116:343–53.
Visentini M, Tinelli C, Colantuono S, Monti M, Ludovisi S, Gragnani L, et al. Efficacy of low-dose rituximab for the treatment of mixed cryoglobulinemia vasculitis: phase II clinical trial and systematic review. Autoimmun Rev [Internet]. 2015;14(10):889–96 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1568997215001214.
Colantuono S, Mitrevski M, Yang B, Tola J, Carlesimo M, De Sanctis GM, et al. Efficacy and safety of long-term treatment with low-dose rituximab for relapsing mixed cryoglobulinemia vasculitis. Clin Rheumatol [Internet]. 2017;36(3):617–23 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28111716.
Stefanutti C, Vivenzio A, Di Giacomo S, Labbadia G, Mazza F, D’Alessandri G, et al. Immunoadsorption apheresis and immunosuppressive drug therapy in the treatment of complicated HCV-related cryoglobulinemia. J Clin Apher [Internet]. 2009;24(6):241–6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19927363.
• Marson P, Monti G, Montani F, Riva A, Mascia MT, Castelnovo L, et al. Apheresis treatment of cryoglobulinemic vasculitis: A multicentre cohort study of 159 patients. Transfus Apher Sci [Internet]. 2018;57(5):639–45 Available from: http://www.ncbi.nlm.nih.gov/pubmed/30228046. An italian retrospective study of 159 patients that suggests the use of apheresis treatment in early life threatening cryoglobulinemic vasculitis.
Puisset F, White-Koning M, Kamar N, Huart A, Haberer F, Blasco H, et al. Population pharmacokinetics of rituximab with or without plasmapheresis in kidney patients with antibody-mediated disease. Br J Clin Pharmacol [Internet]. 2013;76(5):734–40 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23432476.
Terrier B, Marie I, Lacraz A, Belenotti P, Bonnet F, Chiche L, et al. Non HCV-related infectious cryoglobulinemia vasculitis: Results from the French nationwide CryoVas survey and systematic review of the literature. J Autoimmun [Internet]. 2015;65:74–81 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26320984.
Hanbali A, Khaled Y. Incidence of hepatitis B reactivation following Rituximab therapy. Am J Hematol [Internet]. 2009;84(3):195 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19140189.
Yeo W, Chan TC, Leung NWY, Lam WY, Mo FKF, Chu MT, et al. Hepatitis B virus reactivation in lymphoma patients with prior resolved hepatitis B undergoing anticancer therapy with or without rituximab. J Clin Oncol [Internet]. 2009;27(4):605–11 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19075267.
Khan ZH, Ilyas K, Ghazanfar H, Khan HH, Hussain Q, Hammad S, et al. Fatal Fulminant Hepatitis from Rituximab-induced Hepatitis B Reactivation in a Patient with Follicular Lymphoma: A Case Report and a Brief Review of Literature. Cureus [Internet]. 2018;10(3):e2257 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29725560.
Terrier B, Krastinova E, Marie I, Launay D, Lacraz A, Belenotti P, et al. Management of noninfectious mixed cryoglobulinemia vasculitis: data from 242 cases included in the CryoVas survey. Blood [Internet]. 2012;119(25):5996–6004 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22474249.
Schwartz J, Padmanabhan A, Aqui N, Balogun RA, Connelly-Smith L, Delaney M, et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice-Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Seventh Special Issue. J Clin Apher [Internet]. 2016;31(3):149–62 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27322218.
Terrier B, Semoun O, Saadoun D, Sène D, Resche-Rigon M, Cacoub P. Prognostic factors in patients with hepatitis C virus infection and systemic vasculitis. Arthritis Rheum [Internet]. 2011;63(6):1748–57 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21400476.
Landau D-A, Scerra S, Sene D, Resche-Rigon M, Saadoun D, Cacoub P. Causes and predictive factors of mortality in a cohort of patients with hepatitis C virus-related cryoglobulinemic vasculitis treated with antiviral therapy. J Rheumatol [Internet]. 2010;37(3):615–21 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20110523.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
No potential conflicts of interest relevant to this article were reported.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vasculitis
Rights and permissions
About this article

Cite this article
Fuentes, A., Mardones, C. & Burgos, P.I. Understanding the Cryoglobulinemias. Curr Rheumatol Rep 21, 60 (2019). https://doi.org/10.1007/s11926-019-0859-0
Published:
DOI: https://doi.org/10.1007/s11926-019-0859-0