
Abstract
Purpose of Review
A growing body of evidence supports the relevance of the interleukin-23/interleukin-17 (IL-23/IL-17) pathway for the pathogenesis of axial spondyloarthritis (axSpA) and its treatment. Recently, innate lymphoid cells (ILC), a heterogeneous family of immune effector cells, have been identified as a relevant contributor in tissue homeostasis, partially via IL-23/IL-17 axis. This review describes the biology and the origins of the group 3 ILCs (ILC3s) in humans, focusing on their role in the pathogenesis of axSpA.
Recent Findings
Clinical trials showed the effectiveness of IL23/IL-17 axis inhibition in both spondyloarthritis (SpA) and Inflammatory Bowel Disease (IBD). Recent findings confirm the high prevalence of subclinical gut inflammation in patients with SpA. Translational data in humans have demonstrated an increase in the number of ILC3s responsive to IL-23 and producing either IL-22 or IL-17 in the gut of SpA patients. The observation of gut-derived ILC3s in circulation and at inflamed tissues in patients with SpA suggest a recirculation of ILCs from mucosal site to lymphoid tissues and possibly enthesis and joints.
Summary
Multiple observations demonstrate the expansion of IL-17- and IL-22-producing ILC3 in the subclinically inflamed gut of SpA patients. These innate immune cells, also observed in normal entheses, seem to be able to re-circulate from the gut to inflamed tissues of SpA patients, thus contributing to the disease perpetuation. The development of tools that can provide access to diseased tissue from sacroiliac joint and spinal entheses will provide valuable knowledge on the role of ILC3 in axSpA pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The knowledge on the relevance of the interleukin-23-interleukin-17 (IL-23-IL-17) pathway in spondyloarthritis (SpA) pathogenesis has strongly contributed in markedly improving the therapeutic options.
In the last few years, it has become increasingly clear that the main source of IL-17 in patients with SpA is represented by innate immune cells, such as γδ T cell, mucosal-associated invariant T (MAIT) cells, neutrophils, and innate lymphoid cells 3 (ILC3s) [1, 2, 3••, 4, 5••]. Innate lymphoid cells (ILCs) belong to a heterogeneous family of immune effector cells, which have recently gained a great interest in immunology and rheumatology. ILCs are mainly tissue-resident cells, especially situated at epithelial barrier surfaces, where they are involved in early and prompt immune response against pathogens, in tissue remodeling and in maintaining of organ homeostasis [6, 7]. Recent evidence demonstrated that ILCs dysregulation is implicated in the pathogenesis of several inflammatory and immune-mediated diseases [8].
In the intestine, strict cross-talk between microbiota, tissue-resident macrophages and ILC3 producing IL17 and IL-22 seems to be essential for mucosa homoeostasis, and perturbation of this interaction can lead to local and systemic inflammation. Beyond the intestine, ILC3s have been demonstrated to be present in normal human entheses and to be expanded in the peripheral blood and inflamed tissue of patients with ankylosing spondylitis (AS) and those with psoriatic arthritis (PsA). Most of the mechanistic data now available on ILC3 development and biology come however from animal models. This review provides an overview of the most relevant human evidence linking ILC3 with AS and PsA pathogenesis and also highlights the still unanswered questions.
Classification of ILCs and Ontogeny
The earliest identified ILCs belonged to a single group of cytotoxic cells, conventionally named natural killer cells (NK), which are similar and complementary to CD8+ cytotoxic cells, and lymphoid tissue inducer (LTi) cells, that are crucial for the development of lymph nodes during embryogenesis [9, 10]. Works in mice and humans led to the identification of further subtypes by transcription factor expression patterns, cytokine response, and cytokine production.
Based on the most recent data, ILC are further classified into three main groups: (1) conventional NK (cNK), intraepithelial ILC1 (ieILC1), and ILC1; (2) ILC2 group; and (3) ILC3 and LTi cells. However, recent evidence suggests a certain degree of plasticity rather than a terminal differentiation, in line with the ability of these cells to adapt to different micro-environments [11•, 12, 13]. The variegated phenotype, the complex network of transcription factors, and their plasticity make the classification still challenging. Most of the data available today come from mice and are only partially replicated in human. Overall, the classification and the ontogeny of the ILCs remain a developing field and, therefore, caution should be taken in the translation of murine data to humans. However, in both human and mice, the presence and dependence of ILC on the cytokine receptor γc is clear. The γc receptor is a shared component of multiple cytokine receptors including IL-15, IL-2, IL-9, and IL-7. In particular, IL-7 receptor is essential for the development of most of the ILCs as the absence of IL7Rα is associated with altered differentiation of ILCs in both human and mice [14, 15, 16].
In human, NK precursor cells have been found in human fetal liver, bone marrow (BM), cord blood, and adult tonsil [17]. Scoville et al. identified, in secondary lymphoid tissue, a CD34+RORγT+CD45RA+ innate lymphoid tissue precursor (ILCP) with the potential to give rise to all the ILC groups [18]. Similarly, a hematopoietic progenitor cell (HPC) characterized by CD34 positivity and sharing common features with ILC3s has been identified in human tonsil and intestinal lamina propria (LP) [19••]. According to Montaldo et al., these CD34+RORγT− HPCs can potentially still differentiate into both NK and ILC3s whereas CD34+RORγT+ HPC are lineage-specified progenitor of ILC3s [19••] and the fate of HPCs seems to be influenced by the aryl hydrocarbon receptor (AhR) and IL-15 [19••]. Only in 2017, circulating human ILCPs Lin−CD127+CD117+IL1R1+ were discovered in lymphoid and non-lymphoid tissues and were able to differentiate into all ILC subsets. This observation strongly supports the in situ differentiation in response to local signals (pathogens, inflammation, and autoimmunity) [20••].
The transcription factors (TF), involved in the differentiation have been investigated mainly in the mice. The TFs inhibitor of DNA binding 2 (id2), GATA binding protein 3 (Gata3), and nuclear factor IL-3 inducer (Nfil3) have been reported as crucial during the ILC progenitor differentiation in mice [14].
Specifically, Id2 represses the activity of E-box transcription factors (E2A, E2-2, HEB), essential in the early stages of T and B cell development, whereas Gata3 suppresses B cell differentiation, through the block of EBF1 [21,22,23,24,25,26,27]. Both NK and LTi cells require the thymocyte selection-associated HMG box factor (TOX) and Id2 for their differentiation. T-box transcription factor (T-bet) and Eomesodermin (Eomes) characterize the NK cell while LTi cells need the transcription factor RAR-related orphan receptor γt (RORγt) [28,29,30,31,32].
Each population of ILC is characterized by specific transcription factors and associated cytokine expression profile. ILC1, ILC2, and ILC3 reflect the functional polarization and cytokine expression of T helper 1 (Th1), T helper 2 (Th2), and T helper 17 (Th17) cells, respectively, resembling the counterpart of T cells in the innate immune system [31, 33]. Thus, the current classification of ILCs is based on their functional profile and mirrors Th cell nomenclature [34].
Group 1 ILCs
The group 1 ILCs were initially thought to encompass the interferon gamma (IFN-γ)-producing innate cells: conventional NK cells and ILC1s cells [29]. The TF T-Bet is present in the whole group; in addition, the positivity for the TF EOMES and CD103 plus lack of CD127 distinguishes the NK and intraepithelial ILC1 (ieILC1) from other ILC1s [16, 35••]. Group 1 ILCs are mainly tissue-resident cells responsive to interleukin 15 (IL-15), IL-12, and IL-18 and are involved in the primary defense against intracellular pathogens and cancer immuno-surveillance. Similar to Th1 cells, ILC1s promptly secrete tumor necrosis factor alpha (TNFα) and interferon gamma (IFN-γ), but lack significant cytotoxic activity compared to conventional NK (cNK) cells [36]. The ieILC1s have been only recently identified in gut epithelium and tonsils. These cells phenocopy the tissue-resident memory (Trm) cells [37], as are characterized by the production of TGF-beta and the expression of CD103 and CD49a [38].
Group 2 ILCs
ILC2s are tissue-resident cells, featured by the production of type 2 cytokines, such as IL-4, IL-5, IL-13, and IL-9, in response to IL-25, thymic stromal lymphopoietin (TSLP) and IL-33 [35••, 39]. Similar to Th2, ILC2s strictly depend on the high levels of the TF GATA3 [40, 41], and express the chemoattractant receptor expressed on Th2 cells (CRTH2) [42]. ILC2s were initially identified in the human fetal gut tissue and then described also in adipose tissue [43], skin [44], mesenteric lymph nodes, spleen [14], airway, lung, and in Peyer's patches [45]. ILC2s contribute to the first line of defense against helminth and nematodes [46]. In this context, ILC2s limit helminth invasion, through the production of IL-5 and IL-13, which determine the tissue eosinophils recruitment and activation, epithelial and goblet cell hyperplasia followed by increased mucus secretion. [47, 48]. ILC2s are also involved in allergic asthma, chronic sinusitis, nasal polyposis [49], atopic dermatitis [50], and Crohn’s disease [13].
Group 3 ILCs
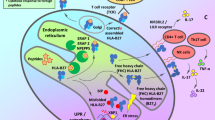
Group 3 ILCs are mainly situated at mucosal barriers, where they are involved in response to the extracellular pathogens (bacteria, fungi) and in maintaining gut homeostasis [51••]. ILC3 consists of two cell types, LTi cells (lymphoid tissue inducer cells) that are important to produce secondary lymphoid organs in embryogenesis and cells designated as NCR+ ILC3 and NCR−ILC3 depending on whether they express NCR receptors of NK cells capable of recognizing pathogenic molecules. [35••, 52]. In human, in fact, the expression of a 44-kD surface molecule, NK cell-associated receptor 44 (NKp44), similar to the natural-cytotoxicity-receptor (NCR) NKp46 observed in mice, distinguishes NKp44+ and NKp44− ILC3s [53]. The cytokine repertoire mirrors this distinction as NKp44+ ILC3s produce IL-22 and the NKp44− ILC3s mainly IL-17, representing respectively the counterpart of Th22 and Th17 [5••, 35••, 54]. ILC3s, in common with the other ILC groups, largely rely on IL-7 stimulation in addition to IL-2 as a signal for survival and proliferation. The activation is mainly controlled by the interaction in situ with the dendritic cells (DC) and CX3CR1 mononuclear phagocytes secreting IL-1 and IL-23, which lead to a prompt induction of IL-17 and IL-22 [11•, 55, 56].
The Lymphoid Tissue inducers (LTi) cells are fundamental for the formation of lymphoid structures; accordingly, LTis were described in the lymph nodes and Peyer’s patches [57], being the most abundant ILCs in the ileum and colon [58]. Only recently, LTi cells have been included in group 3 ILC for the high similarity with the ILC3, including the production of IL-17 [59]. In particular, the expression of neuropilin (NRP-1) is limited to LTi and usually is absent in NKp44− ILC3s [60]. Recent evidence, suggest a role of LTi in the formation of ectopic lymphoid structures associated with chronic inflammation [60]. Studies in animal demonstrated the initial appearance of LTi type of ILC3 early in the gut during embryogenesis with the absence of ILC3 NKp46+. Only after birth, they proliferate outnumbering the other ILC3 subsets [61]. On the light of this observation, a role for the environmental factor in driving the expansion of ILC3 has been demonstrated: microbiota and diet recognized as two relevant players [62]. The qualitative and quantitative alteration has suggested a bidirectional relationship between ILC3 and the gut microbiota in animal model lacking ILC3 [63].
ILC3s have a role in the compartmentalization of the microbiota as the lack of ILC3s was associated with immunization toward commensal bacteria due to a breach in the intestinal epithelial barrier [64]. On the other hand, commensal bacteria and related products foster the activation and expansion of ILC3s in the gut [61, 65]. It is worth noting that ILC3s lack pattern recognition receptors (PRR) such as Toll-like receptors that are able to sense microbial products. Therefore, an intricate network of intercellular communication and soluble mediators is in place to activate ILC3 in the presence of danger signals. In particular, dendritic cells and macrophages have been identified as a major source of IL-23, which stimulates and activates ILC3s. In this regard, a specific population of intestinal mononuclear phagocytes (MNP) CX3CR1+ has been suggested as a bridge immune response from the intestinal lumen to the innate immune system [56]. More recently, it has been demonstrated that ILC3s regulate steady-state interactions between T follicular helper cells (Tfh) and B cells to limit mucosal IgA responses. ILC3s use conserved migratory cues to establish residence within the interfollicular regions of the intestinal draining lymph nodes, where they act to limit Tfh responses and B cell class switching through antigen presentation. The absence of ILC3-intrinsic antigen presentation results in increased and selective IgA coating of bacteria residing within the colonic mucosa, implicating lymph node resident, antigen-presenting ILC3s as a critical regulatory checkpoint in the generation of T cell-dependent colonic IgA and in maintaining tissue homeostasis and mutualism with the mucosal-dwelling commensal microbiota [66]. Mediators other than cytokines are involved in controlling ILC3 homeostasis and activation. Short-chain fatty acids (SCFA), in particular butyrate produced by bacterial metabolism, directly regulate ILC3s [67]. In addition, AhR ligands and vitamin A derived retinoids produced by bacterial metabolism stimulate ILCs differentiation, specifically the maturation of LTi cells and ILC3s in the adult small intestine [68, 69].
The bone marrow (BM), secondary lymphoid organs, and gut are known niches for ILC3s development, expansion, and priming; however, less is known about ILC recirculation migration programs. As stressed before, the local chemokine and cytokine milieu may be a key factor for ILC3 recruitment and expansion in situ. Recent work demonstrated both the presence of NKp46+ILC3 and ILCPs in circulation. Those cells are differently expressing α4β7, CCR7, CCR9, and CXCR6 with a known role in tissue tropism [70]. In the lung, ILC3 producing IL17A could foster the formation of ectopic lymphoid structures during chronic inflammation [60]. Similarly, the known role of IL-23/IL17 axis in inflammatory condition, such as psoriasis, IBD, and arthritis, suggests the involvement of IL-22 and IL-17 producing ILC3s in these conditions.
Ankylosing Spondylitis Associated Subclinical Gut Inflammation
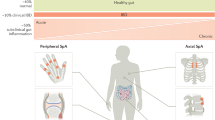
The strict correlation between gut inflammation and SpA is well characterized. Its description dates back to 1976, with the analysis to the frequency of joint involvement in the course of inflammatory bowel diseases (IBD) [71]. The development of inflammatory arthritis occurs in nearly half of the patients with a diagnosis of IBD. On the other hand, up to 60% of ankylosing spondylitis (AS) patients show histologic sign of subclinical gut inflammation. Among those, only 10% will progress to a clinically and histologically defined diagnosis of IBD [72]. Analysis performed on the Gent Inflammatory arthritis, and spondylitis cohort (GIANT) revealed a correlation between the gut inflammation and the BM edema in sacroiliac joints [73•]. Recently, flow cytometry analysis showed a distinct immune signature in the colon of patients affected by AS compared to IBD in the absence of inflammation, supporting the existence of a SpA-specific gut inflammation [74•].
The gut inflammation in SpA has been classified into two main histologic presentations: acute and chronic type of inflammation. The acute type is featured by bacterial enterocolitis and increased expansion of mononuclear cells in the lamina propria [75]. The chronic type of inflammation is characterized by crypts distortion, villous atrophy, and mononuclear cell infiltration of the lamina propria lymphoid follicles. Consensual hyperplasia of goblet cells, increased mucin production, and Paneth cells activation have also been observed [76, 77].
Recent works link the subclinical gut inflammation to a certain degree dysbiosis, defined as a qualitative and quantitative perturbation in gut microbiota. Metagenomic studies involving the sequencing of the 16S ribosomal RNA gene isolated from stool samples and culture of intestinal biopsies demonstrated a significant expansion of some bacterial families, such as Raminococcus, Dialister, Prevotella, and Porphyromonas, and a relative reduction in Bacteroides species [78, 79, 80].
Our group previously demonstrated that the local inflammation and the dysbiosis are associated with the damage in the gut-vascular and gut epithelial barrier [81]. Translocation of bacterial products was observed in the submucosa and circulating blood, confirming the presence of a so-called leaky gut [81]. The altered intestinal permeability has been linked to the derangement of the epithelial tight-junction and higher levels of zonulin, a cellular junction modulator [81].
There is still discussion and need for further translational studies on the causal trigger of these alterations. Host-related factors such as genetics may influence the interaction between the microbiome and immune system at mucosal sites. Experiments in Lewis rats transgenic for HLA-B27 revealed a difference in cecal microbiota compared to the wild type [82, 83]. HLA-B27 is a major histocompatibility complex class I molecule with a role in preventing viral and bacterial antigens. HLA-B27 is the strongest risk factor for axSpA but not for IBD; however, common genetic predisposing factors other than HLA-B27 that are shared between AS and IBD have been identified [84]. On the other hand, we reported that bacteria isolated from the ileum of SpA patients could up-regulate ex vivo the production of zonulin from cultured cells, in support of a direct role of dysbiosis in the pathogenesis of gut inflammation in axSpA [85].
ILC3 Expansion and Recirculation in Ankylosing Spondylitis
Our previous works demonstrated the expansion of IL-17 and IL-22 producing ILC3s in the gut of patients affected by AS [5••]. The presence of these cells was also observed in peripheral blood, bone marrow, and synovial fluids, and it correlated with the Bath Ankylosing Spondylitis Disease Activity Index [5••]. On the light of these observations, the gut has been proposed as a possible base camp for ILC3 differentiation, expansion, and priming in AS [86]. Lyn−NKp44+T-bet+RORγt−IL23R+ILC3s were particularly expanded in the gut of AS patients with acute or chronic gut inflammation only and were associated with increased mucins production and goblet cells hyperplasia [5••]. Specifically, the cryptopatches in lamina propria rich in LTi aggregates are sites of ILC3 maturation in the gut and cryptopatches-like structures have been observed in the gut of AS patients [5••, 87].
Moreover, in patients with AS, ILC3s were also found to be expanded in the peripheral blood, synovial fluid, and bone marrow (BM), and to express the homing integrin α4β7 [5••]. In the peripheral blood, ILC3s prevalently produced IL-22, and the production of IL-17 alone or IL-22/IL17 confined to a small subset of these cells. Among synovial fluid and BM mononuclear cells, ILC3 produced IL-22 exclusively. Besides, the mucosal vascular cell adhesion molecule 1 (MADCAM-1), the α4β7 ligand, was found to be significantly up-regulated in the gut and in the inflamed BM of patients with AS, suggesting a recirculation of ILC3 from gut to the inflamed AS tissues [5••]. In this context, inflammatory tissues rich in MADCAM-1, IL-23, and IL-7 could be site of expansion and migration of ILC3.
Role of CX3CR1+ Mononuclear Phagocytes in Supporting ILC3 Expansion via IL-23
Intestinal mononuclear phagocytes (MNPs) are strategically positioned in the gut-associated lymphoid tissue (GALT) and in the subepithelial lamina propria [88]. In mice, lamina propria resident MNPs can be subdivided based on their surface expression of the integrin CD103 or CX3 C-chemokine receptor 1 (CX3CR1+). These subpopulations of MNPs differ in terms of their ability to take up antigen and trigger intestinal T and B cell response. CX3CR1+ MNPs are thought to be conventional myeloid dendritic cells (DC) as they develop from a classical DC precursor [89], express CCR7, migrate to the mesenteric lymph nodes (MLNs) upon TLR stimulation, and can activate naïve T cells in vitro [90, 91]. CX3CR1+ lamina propria cells do not derive from the classic DC precursors but are the progeny of Ly-6Chi circulating monocytes [88]. CX3CR1+ MNPs can be further divided into cells expressing high and intermediate levels of the chemokine receptor. CX3CR1+ MNPs were shown to modulate adaptive immunity, through the promotion of Th17 cell differentiation [92] and FOXP3 maintenance on T reg cells [93]. Even if these cells were previously thought to be tissue-resident and non-migratory, recent data reported that during intestinal inflammation, CX3CR1+ MNPs can up-regulate CCR7 and acquire the ability to migrate to secondary lymphoid organs [94]. At steady state, the microbiota inhibit the transport of commensal bacteria from the lumen to the MLNs. However, under conditions of dysbiosis, non-invasive bacteria were trafficked by CX3CR1+ MNPs to the MLNs and induced T cell responses. In naïve SKG mouse intestine characterized by intestinal dysbiosis, CX3CR1+ MNPs are significantly expanded [95•]. These findings suggest a central role for microbiota in regulating the antigen access from the intestinal lumen to the MLNs by a CX3CR1+ cell-mediated pathway. In predisposed subjects, CX3CR1+ MNPs can bridge immune response from the intestinal lumen to the innate immune system systemically.
Confirming this hypothesis, recent evidence reported that the intestinal replenishment of CX3CR1+ MNPs derives from Ly-6Chi monocytes and, under inflammatory conditions, Ly6Chi monocytes accumulate in secondary lymphoid organs in a DcR-6 dependent manner [96, 97]. DcR-6 is a scavenger receptor which acts as a negative regulator of inflammation, driving degradation of majority of the inflammatory CC chemokines. The control of CC chemokines by DcR-6 seems to have a particular impact on the traffic Ly6Chi monocytes. In DcR-6-deficient mice, there is a delay in the clearance of inflammatory CC chemokines which promote, under inflammatory conditions, an increased concentration of CCL2, and significant recruitment of Ly-6Chi MNPs in secondary lymphoid organs [97]. Thus, the CX3CR1+-mediated immune response seems to be affected by a balance between CCL2 and DcR-6 effects.
Similar data were observed in AS patients, where a significantly decreased expression of DcR-6 in the gut, BM, and synovial samples have been reported [95•]. Moreover, DcR-6 deficiency correlated with an increase in tissue and systemic expression of CCL2 as well as an increase in CX3CR1+ CD59+ cells (CD59 is the human homolog of Ly6). Pro-inflammatory CX3CR1+ CD59+, but not resident CX3CR1+ CD59− cells were expanded in the gut of AS patients compared to controls and correlated with the presence of bacteria. Transcriptome analysis revealed that CX3CR1+ CD59+ cells showed a specific pro-inflammatory signature in AS patients. Inflammatory CX3CR1+ MNPs were also expanded in the peripheral blood and inflamed tissues of AS patients [95•]. The majority of these cells expressed the marker of intestinal homing CCR9 [98] and produce high levels of IL-23 and TNF-like molecule 1 A (TL1A) [95•]. Interestingly, the frequency of circulating CX3CR1+CD59+CCR9+IL-23+TL1A+ monocytes significantly correlated with the frequency of circulating ILC3 as well as with disease activity. In the presence of isolated CX3CR1+ CD59+ MNPs from the gut and peripheral blood of AS patients, significant activation of ILC3 producing IL-22 was observed, indicating an important role of these cells in supporting ILC3 activation and expansion [95•]. These data suggest that in AS patients, CX3CR1+CD59+ monocytes may acquire a pro-inflammatory phenotype, migrating from the gut to inflamed tissues and actively participating in the activation and expansion of ILC3.
ILC3 in Enthesis
The correlation demonstrated between the ILC3 and the disease activity score in AS, in addition to the strong role of IL-23, IL-22, and IL17A in the pathogenesis of AS, point to ILC3 as a possible culprit in the AS pathogenesis and disease progression. Recently, a subset of T cells highly responsive to IL-23 has been observed in the entheses of a murine model of AS [99]. Interestingly, these murine enthesial T cells shared several immunological similarities with ILC3. These cells are Lyn−, express the IL-23R and release IL-17 and IL-22, and important in driving inflammation [99]. However, the presence of ILC3 in human spinal entheses has been demonstrated only recently. McGonagle’s group examined the interspinous ligament and spinous process bone of healthy individuals undergoing spinal surgery [100•]. ILC3 cells were detected by flow cytometry and expression of RORC by qRT-PCR. The study revealed a higher percentage of NKp44+ ILC3 in enthesiaI soft tissue and peri-enthesial bone. Accordingly, increased expression of IL-23 receptor was observed in these cells, supporting the role of IL-23 also at this site [100•].
ILC3 in Other Spondyloarthritides
Recent study results have analyzed the ILC3 role in psoriatic arthritis (PsA) [101, 102]. In particular, Soare et al. recently demonstrated that PsA is characterized by alterations of circulating ILCs. In clinically active PsA, ILC3s were significantly increased at the expense of ILC2s and the ILC2/3 ratio correlated with both composite clinical disease activity scores, but also with imaging signs of inflammation and structural damage, supporting the concept of IL-23/IL-17 pathway activation as the driving force of inflammation in PsA patients [101]. Leijten and co-workers also demonstrated the expansion of ILC3 in the synovial fluids of PsA patients but not in peripheral blood [102]. In particular, fewer CCR6+ ILCs were found in PsA peripheral blood (PB) than in healthy control PB, while PsA synovial fluid was enriched for CCR6+ ILCs compared to PsA PB and RA synovial fluid. Increased numbers of IL-17A-producing ILC3 were present in PsA synovial fluids compared to RA synovial fluids. CCR6, NKp44, and melanoma cell adhesion molecule (MCAM) were expressed on the cell surface of synovial fluid ILC3s and the number of circulating NKp44+, CCR6+, and MCAM+ ILCs in blood was inversely correlated with PsA disease activity.
Conclusions
There is solid evidence suggesting a pathogenic role of innate lymphoid cells and specifically ILC3s in axSpA as summarized in Fig. 1. In particular, ILC3 are significant producers of the inflammatory cytokine IL-17 in response to IL-23. The inhibition of the IL-23/IL-17 axis has been groundbreaking in the treatment of SpA. However, the available data fail to identify causes and consequences. The intestinal dysbiosis observed in the course of AS, could trigger the activation and expansion of ILC3s and possibly trigger the articular involvement. Germ-free SKG mice are resistant to the development of SpA manifestations [103, 104]. Moreover, dysregulated ILC3s can per se lead to qualitative and quantitative alteration of the commensal flora. Conversely, recent studies in mice have demonstrated that IL-23 itself could favor the outgrowth of SpA-associated pathobionts (potentially pathological organisms which, under normal circumstances, live as a symbionts) and suppress host support for homeostatic microbiota [105]. Similarly, in humans, the TNF-blockade could lead to a reduction in the dysbiotic bacteria and candidate arthritogenic bacterial peptides [106]. Further studies are needed to elucidate mechanistically the causes of ILC3s expansion in axSpA and the impact of therapeutic manipulation of the intestinal microbiota in this disease. Multiple clues suggest the recirculation of ILC3s from the gut to the entheses; however, translational studies investigating human spinal entheses and sacroiliac joints are warranted.

Subclinical gut inflammation observed in axial-SpA is associated to dysbiosis, goblet cell hyperplasia, and altered permeability intestinal epithelial barrier. Deranged bacteria compartmentalization and increased invasiveness lead to bacterial translocation in the submucosa. Bacterial products, AhR ligands, and vitamin A derived retinoids produced by bacterial metabolism stimulate ILCs differentiation while short-chain fatty acids (SCFA) inhibit ILC3expansion. ILC3 undergo maturation and priming in cryptopatches rich in LTi within the lamina propria and supported by CX3CR1+CD59+ intestinal mononuclear phagocytes. In response to IL-23, the expanded ILC3 produce IL17 and IL-22 contributing to gut inflammation. Recirculation of ILC3 between bone marrow, secondary lymphoid organs, skin, synovium, and enthesis could participate in the articular manifestations of SpA
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum [Internet]. 2012;64:1420–9. https://doi.org/10.1002/art.33507.
Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis. 2016;75:2124–32.
•• Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL-17+ cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther [Internet]. 2011;13:R95. https://doi.org/10.1186/ar3370. This report demonstrated the prevalence of lL-17 positive cell other than Th17 in spinal facet joints of patients with AS.
Noordenbos T, Yeremenko N, Gofita I, van de Sande M, Tak PP, Caňete JD, et al. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum [Internet]. 2012;64:99–109. https://doi.org/10.1002/art.33396.
•• Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis [Internet]. 2015;74:1739–47. https://doi.org/10.1136/annrheumdis-2014-206323. This study demonstrated the expansion of gut-derived lL-17+ and lL-22+lLC3s, supporting gut/joint axis theory.
Mjösberg J, Spits H. Human innate lymphoid cells. J Allergy Clin Immunol. 2016;138:1265–76.
Neill DR, Flynn RJ. Origins and evolution of innate lymphoid cells: wardens of barrier immunity. Parasite Immunol. 2018;40:e12436.
McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41:366–74.
Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5:117–21.
Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTβ+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493–504.
• Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127+ group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. 2015;43:146–60. Work demonstrating the plasticity of intestinal lLCs controlled by lL-12 and lL-23 in inflamed gut.
Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting edge: notch signaling promotes the plasticity of group-2 innate lymphoid cells. J Immunol. 2017;198:1798–803.
Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213:569–83.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature. 2010;463:540–4.
Vonarbourg C, Diefenbach A. Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Semin Immunol. 2012;24:165–74.
Nagasawa M, Spits H, Ros XR. Innate lymphoid cells (ILCs): cytokine hubs regulating immunity and tissue homeostasis. Cold Spring Harb Perspect Biol. 2017;10:a030304.
Renoux VM, Zriwil A, Peitzsch C, Michaëlsson J, Friberg D, Soneji S, et al. Identification of a human natural killer cell lineage-restricted progenitor in fetal and adult tissues. Immunity. 2015;43:394–407.
Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, et al. A progenitor cell expressing transcription factor RORγt generates all human innate lymphoid cell subsets. Immunity. 2016;44:1140–50.
•• Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, et al. Human RORγt+CD34+ cells are lineage-specified progenitors of group 3 RORγt+ innate lymphoid cells. Immunity. 2014;41:988–1000. Seminal work that identified human progenitor cells committed precursor of lLC3s.
•• Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. 2017;168:1086–1100.e10. Seminal paper demonstrating circulating progenitor and supporting the theory of in situ “lLC-poiesis”.
Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue–inducing cell development requires Id2-mediated suppression of E protein activity. J Exp Med. 2007;204:1119–30.
Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157:340–56.
Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508:397–401.
Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORγt sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med. 2012;209:729–40.
Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med. 2014;211:1733–40.
Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med. 2014;211:1723–31.
Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity. 2014;40:378–88.
Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67.
Harmon C, Robinson MW, Fahey R, Whelan S, Houlihan DD, Geoghegan J, et al. Tissue-resident Eomes hi T-bet lo CD56 bright NK cells with reduced proinflammatory potential are enriched in the adult human liver. Eur J Immunol. 2016;46:2111–20.
Collins A, Rothman N, Liu K, Reiner SL. Eomesodermin and T-bet mark developmentally distinct human natural killer cells. JCI Insight. 2017;2:e90063.
Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–7.
Naito T, Shiohara T, Hibi T, Suematsu M, Ishikawa H. RORγt is dispensable for the development of intestinal mucosal T cells. Mucosal Immunol. 2008;1:198–207.
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells — a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–9.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–66.
•• Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. 2017;46:148–61. This report provides a deep phenotyping of lLC subsets via mass cytometry.
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol [Internet]. 2013;14:221–9. https://doi.org/10.1038/ni2534.
Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. 2018;283:54–76.
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity. 2013;38:769–81.
Camelo A, Rosignoli G, Ohne Y, Stewart RA, Overed-Sayer C, Sleeman MA, et al. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv [Internet]. 2017;1:577 LP–589. https://doi.org/10.1182/bloodadvances.2016002352.
Hoyler T, Klose CSN, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37:634–48.
Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37:649–59.
Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–62.
Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519:242–6.
Spits H. Group 2 innate lymphoid cells show up in the skin. Immunol Cell Biol. 2013;91:390–2.
Hashiguchi M, Kashiwakura Y, Kojima H, Kobayashi A, Kanno Y, Kobata T. Peyer’s patch innate lymphoid cells regulate commensal bacteria expansion. Immunol Lett. 2015;165:1–9.
Koyasu S, Moro K, Tanabe M, Takeuchi T. Natural Helper Cells. 2010:21–44.
Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43:29–40.
Price AE, Liang H-E, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci. 2010;107:11489–94.
Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria J-P, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137:75–86.e8.
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210:2939–50.
•• Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell [Internet] Elsevier Inc. 2017;171:201–216.e18. https://doi.org/10.1016/j.cell.2017.07.027. Identification of lL-10 producing regulatory lLCs termed lLCreg.
Ciccia F, Accardo-Palumbo A, Alessandro R, Rizzo A, Principe S, Peralta S, et al. Interleukin-22 and interleukin-22-producing NKp44+ natural killer cells in subclinical gut inflammation in ankylosing spondylitis. Arthritis Rheum. 2012;64:1869–78.
Hoorweg K, Peters CP, Cornelissen F, Aparicio-Domingo P, Papazian N, Kazemier G, et al. Functional differences between human NKp44(−) and NKp44(+) RORC(+) innate lymphoid cells. Front Immunol. 2012;3:72. https://doi.org/10.3389/fimmu.2012.00072.
Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, et al. IL-18 drives ILC3 proliferation and promotes IL-22 production via NF-κB. J Immunol [Internet]. 2017;199:ji1601554. https://doi.org/10.4049/jimmunol.1601554.
Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9.
Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med [Internet]. 2014;211:1571–83. https://doi.org/10.1084/jem.20140678.
Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity [Internet]. 1997;7:493–504. https://doi.org/10.1016/S1074-7613(00)80371-4.
Forkel M, Mjösberg J. Dysregulation of group 3 innate lymphoid cells in the pathogenesis of inflammatory bowel disease. Curr Allergy Asthma Rep. 2016;16:73.
Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74.
Shikhagaie MM, Björklund ÅK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 is expressed on lymphoid tissue residing LTi-like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep. 2017;18:1761–73.
Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORγt + innate lymphoid cells. Science. 2010;330(80):665–9.
Constantinides MG. Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol. 2018;103:409–19.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J Exp Med. 2011;208:125–34.
Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–70.
Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, et al. Antigen-presenting ILC3 regulate T cell–dependent IgA responses to colonic mucosal bacteria. J Exp Med [Internet]. 2019;216:jem.20180871. https://doi.org/10.1084/jem.20180871.
Kim S-H, Cho B-H, Kiyono H, Jang Y-S. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep. 2017;7:3980.
van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508:123–7.
Li S, Bostick JW, Zhou L. Regulation of innate lymphoid cells by aryl hydrocarbon receptor. Front Immunol. 2018;8. https://doi.org/10.3389/fimmu.2017.01909.
Kim CH, Hashimoto-Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016;37:68–79.
Deshayes P, Houdent C, Hecketsweiler P. Rheumatic manifestations of Crohn’s disease. A national survey. Rev Rhum Mal Osteoartic. 1976;43:541–51.
De Vos M, Mielants H, Cuvelier C, Elewaut A, Veys E. Long-term evolution of gut inflammation in patients with spondyloarthropathy. Gastroenterology. 1996;110:1696–703.
• Van Praet L, Jans L, Carron P, Jacques P, Glorieus E, Colman R, et al. Degree of bone marrow oedema in sacroiliac joints of patients with axial spondyloarthritis is linked to gut inflammation and male sex: results from the GIANT cohort. Ann Rheum Dis. 2014;73:1186–9. This clinical study demonstrated the association between gut and joint inflammation in a large cohort.
Dunn ETJ, Taylor ES, Stebbings S, Schultz M, Butt AG, Kemp RA. Distinct immune signatures in the colon of Crohn’s disease and ankylosing spondylitis patients in the absence of inflammation. Immunol Cell Biol. 2016;94:421–9.
Mielants H, Veys EM, Cuvelier C, De Vos MD. Ileocolonoscopic findings in seronegative spondylarthropathies. Rheumatology. 1988;XXVII:95–105. https://doi.org/10.1093/rheumatology/XXVII.supp_2.95.
Ciccia F, Bombardieri M, Rizzo A, Principato A, Giardina AR, Raiata F, et al. Over-expression of paneth cell-derived anti-microbial peptides in the gut of patients with ankylosing spondylitis and subclinical intestinal inflammation. Rheumatology. 2010;49:2076–83.
De Vos M, Cuvelier C, Mielants H, Veys E, Barbier F, Elewaut A. Ileocolonoscopy in seronegative spondylarthropathy. Gastroenterology. 1989;96:339–44.
Tito RY, Cypers H, Joossens M, Varkas G, Van Praet L, Glorieus E, et al. Brief report: dialister as a microbial marker of disease activity in spondyloarthritis. Arthritis Rheum. 2017;69:114–21.
Stoll ML, Kumar R, Morrow CD, Lefkowitz EJ, Cui X, Genin A, et al. Altered microbiota associated with abnormal humoral immune responses to commensal organisms in enthesitis-related arthritis. Arthritis Res Ther. 2014;16:486.
Costello M-E, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, et al. Brief report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol [Internet]. 2015;67:686–91. https://doi.org/10.1002/art.38967.
Ciccia F, Guggino G, Rizzo A, Alessandro R, Luchetti MM, Milling S, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis [Internet]. 2017;76:1123–32. https://doi.org/10.1136/annrheumdis-2016-210000.
Lin P, Bach M, Asquith M, Lee AY, Akileswaran L, Stauffer P, et al. HLA-B27 and human β2-microglobulin affect the gut microbiota of transgenic rats. Bereswill S, editor. PLoS One. 2014;9:e105684. https://doi.org/10.1371/journal.pone.0105684.
Gill T, Asquith M, Brooks SR, Rosenbaum JT, Colbert RA. Effects of HLA–B27 on gut microbiota in experimental spondyloarthritis implicate an ecological model of dysbiosis. Arthritis Rheum. 2018;70:555–65.
Thjodleifsson B, Geirsson ÁJ, Björnsson S, Bjarnason I. A common genetic background for inflammatory bowel disease and ankylosing spondylitis: a genealogic study in Iceland. Arthritis Rheum. 2007;56:2633–9.
Ciccia F, Guggino G, Rizzo A, Alessandro R, Luchetti MM, Milling S, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis. 2017;76:1123–32.
Neerinckx B, Elewaut D, Lories RJ. Spreading spondyloarthritis: are ILCs cytokine shuttles from base camp gut? Ann Rheum Dis. 2015;74:1633–5.
Eberl G, Sawa S. Opening the crypt: current facts and hypotheses on the function of cryptopatches. Trends Immunol. 2010;31:50–5.
Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol. 2010;10:415–26.
Varol C, Landsman L, Jung S. Probing in vivo origins of mononuclear phagocytes by conditional ablation and reconstitution. Methods Mol Biol. 2009;531:71–87.
Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, et al. Intestinal CD103 + , but not CX3CR1 + , antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med. 2009;206:3101–14.
Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–25.
Denning TL, Wang Y, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat Immunol. 2007;8:1086–94.
Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–84.
Diehl GE, Longman RS, Zhang J-X, Breart B, Galan C, Cuesta A, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature. 2013;494:116–20.
• Ciccia F, Guggino G, Zeng M, Thomas R, Ranganathan V, Rahman A, et al. Proinflammatory CX3CR1+CD59+tumor necrosis factor–like molecule 1A+interleukin-23+ monocytes are expanded in patients with ankylosing spondylitis and modulate innate lymphoid cell 3 immune functions. Arthritis Rheum. 2018;70:2003–13. This work demonstrated the expansion of lL23 producing CX3CR1+ mononuclear phagocytes, supporting lLC3 expansion in the gut of AS patients.
Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15:929–37.
Savino B, Castor MG, Caronni N, Sarukhan A, Anselmo A, Buracchi C, et al. Control of murine Ly6Chigh monocyte traffic and immunosuppressive activities by atypical chemokine receptor D6. Blood. 2012;119:5250–60.
Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, et al. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci. 2007;104:6347–52.
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat Med. 2012;18:1069–76.
•• Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: group 3 innate lymphoid cells in human enthesis. Arthritis Rheum. 2017;69:1816–22. Unique report investigating the presence of lLC3 in human spinal enthesis.
Soare A, Weber S, Maul L, Rauber S, Gheorghiu AM, Luber M, et al. Cutting edge: homeostasis of innate lymphoid cells is imbalanced in psoriatic arthritis. J Immunol [Internet]. 2018;200:1249–54. https://doi.org/10.4049/jimmunol.1700596.
Leijten EFA, van Kempen TS, Boes M, Michels-van Amelsfort JMR, Hijnen D, Hartgring SAY, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol (Hoboken, NJ). 2015;67:2673–8.
Ruutu M, Thomas G, Steck R, Degli-Esposti MA, Zinkernagel MS, Alexander K, et al. β-glucan triggers spondylarthritis and Crohn’s disease-like ileitis in SKG mice. Arthritis Rheum [Internet]. 2012;64:2211–22. https://doi.org/10.1002/art.34423.
Rehaume LM, Mondot S, Aguirre de Cárcer D, Velasco J, Benham H, Hasnain SZ, et al. ZAP-70 genotype disrupts the relationship between microbiota and host, leading to spondyloarthritis and ileitis in SKG mice. Arthritis Rheumatol [Internet]. 2014;66:2780–92. https://doi.org/10.1002/art.38773.
Rehaume LM, Matigian N, Mehdi AM, Lachner N, Bowerman KL, Daly J, et al. IL-23 favours outgrowth of spondyloarthritis-associated pathobionts and suppresses host support for homeostatic microbiota. Ann Rheum Dis [Internet]. 2019;78:494–503. https://doi.org/10.1136/annrheumdis-2018-214381.
Yin J, Sternes PR, Wang M, Morrison M, Song J, Li T, et al. Shotgun metagenomics reveals an enrichment of potentially cross-reactive bacterial epitopes in ankylosing spondylitis patients, as well as the effects of TNFi therapy and the host’s genotype upon microbiome composition. bioRxiv [Internet]. 2019;571430. https://doi.org/10.1101/571430.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Spondyloarthritis
Rights and permissions
About this article

Cite this article
Mauro, D., Macaluso, F., Fasano, S. et al. ILC3 in Axial Spondyloarthritis: the Gut Angle. Curr Rheumatol Rep 21, 37 (2019). https://doi.org/10.1007/s11926-019-0834-9
Published:
DOI: https://doi.org/10.1007/s11926-019-0834-9