
Abstract
Purpose of Review
A new autoinflammatory disease, deficiency of adenosine deaminase 2 (DADA2), caused by mutations in the CECR1 gene, was first reported in 2014. This review aims to update progress in defining, treating, and understanding this multi-faceted disorder.
Recent Findings
DADA2 was first described in patients with systemic inflammation, mild immune deficiency, and vasculopathy manifested as recurrent stroke or polyarteritis nodosa (PAN). More than 125 patients have now been reported, and the phenotype has expanded to include children and adults presenting primarily with pure red cell aplasia (PRCA), or with antibody deficiency. Age of onset and clinical severity vary widely, even among related patients, and are not clearly related to CECR1 genotype. Inflammatory features often respond to anti-TNF agents, but marrow failure and severe immune deficiency may require hematopoietic stem cell transplantation.
Summary
ADA2 is expressed and secreted by monocytes and macrophages, but its biological function and the pathogenesis of DADA2 are uncertain and will remain an important area of research. Pre-clinical investigation of ADA2 replacement therapy and CECR1-directed gene therapy are warranted, but complicated by the absence of a suitable animal model.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genome-wide association studies (GWAS) have identified many loci that influence the inflammatory and autoimmune diseases most familiar to rheumatologists, but their diagnosis relies on clinical presentation, pathological findings, and disease-associated autoantibodies. By contrast, gene sequencing is required for unambiguous diagnosis of more than 25 rare monogenic autoinflammatory diseases, which typically lack specific biomarkers [1]. Most of these genes were identified by positional cloning, homozygosity mapping, and candidate gene screening of large pedigrees. In the last few years, whole-exome sequencing (WES) has enabled unbiased discovery of mutated genes in single or small numbers of patients with novel clinical presentations.
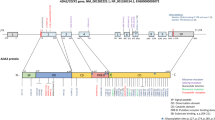
In 2014, investigators at the NIH and in Israel independently reported the use of WES to define a new autoinflammatory disorder caused by loss of function mutations in both copies of CECR1 (cat eye syndrome critical region candidate 1). This 10-exon/28-kb gene on chromosome 22 q11.1 encodes a 511-amino-acid protein, adenosine deaminase 2 (ADA2); the disorder has been named deficiency of ADA2, abbreviated DADA2 [2••, 3••]. ADA2 was discovered 40 years ago in tissues of patients with severe combined immune deficiency (SCID) who lacked ADA1, the major ADA activity in humans, which is encoded by the ADA gene on chromosome 20 [4]. ADA1 is a largely intracellular, 41-kDa monomer expressed in all cells, with highest levels in lymphocytes. ADA2 is a homodimer of 57-kDa subunits; it is expressed mainly in myeloid cells and is secreted by monocytes, macrophages, and dendritic cells [5, 6]. Structurally, the proteins have similar active sites, but ADA2 has additional domains involved in its secretion, dimerization, and binding to a putative receptor [7, 8]. Mice lack a CECR1 orthologue and do not express ADA2.
The initial report of ADA2 showed that it had much lower affinity for its substrates, adenosine (Ado) and 2′-deoxyadenosine (dAdo), than does ADA1, and is insensitive to a purine analog, EHNA, which strongly inhibits ADA1 [4]. Assays based on this differential inhibition have shown that ADA2 activity is elevated in serum of patients with chronic hepatitis and some infections, including tuberculosis and HIV, as well as from patients with various primary immunodeficiency diseases [9]. Though intriguing, this finding has had little utility, and only in retrospect does it hint at some critical function of ADA2 in humans. Assay of plasma ADA2 activity now provides an inexpensive and rapid means of testing for DADA2.
Clinical Presentation of the First DADA2 Patients
The first two reports of DADA2 described 33 patients with periodic fever, livedoid rash, elevated inflammatory markers, vasculopathy, and mild immune deficiency mainly affecting B cells [2••, 3••]. Vasculopathy was manifested primarily as early-onset lacunar stroke in one study, and as polyarteritis nodosa (PAN) in the other, reflecting the presentation of the index patients. Thirteen different CECR1 mutations were identified, mostly missense, which mapped to the signal peptide, catalytic, dimerization, and putative receptor binding domains of ADA2. In addition to showing that DADA2 is clinically and genetically heterogeneous, these reports provided important insight into pathogenesis and therapy (for other recent reviews, see [10, 11]).
Of the nine NIH patients, eight had strokes, in most cases, by age 5 years, which were ischemic, hemorrhagic, or both, and on neuroimaging, localized to deep nuclei, midbrain, and brain stem [2••]. The 9th patient had small vessel vasculitis. In addition to livedo racemosa and fever (8/9 patients), other findings in four or five patients each included hepatosplenomegaly, hypogammaglobulinemia (with recurrent bacterial and viral infections in two of four patients), variable lymphopenia, and low serum IgM. All were ANCA-negative. Skin biopsies showed vasculitis of medium-sized arteries. Brain biopsies in two patients showed erythrocyte extravasation without inflammation. Endothelial damage and endothelial cell activation were also found in skin and brain biopsies, along with increased staining for IL-1β, iNOS, and TNF-α. Plasma levels of these and other cytokines were normal. In several patients, circulating memory B cells were reduced, and B cells showed increased apoptosis in vitro. The activity of ADA1 in red cells was normal, and there was no elevation in dAdo nucleotides (a hallmark of ADA1 deficiency). There was no sustained clinical response to immunosuppressive therapy, or to anakinra, an inhibitor of IL-1 that is effective in several other autoinflammatory diseases.
Elkan et al. described 24 patients with features of PAN, including 19 from 5 Israeli families of Georgian Jewish origin, who had been followed for many years; 4 German patients from a single family; and 1 Turkish patient (of 14 with PAN in whom CECR1 was sequenced) [3••]. The patients were negative for ANCA and had histopathologic findings consistent with PAN [12]. Notably, disease severity and age of onset were highly variable, even within families. Four patients had only cutaneous PAN (cPAN) without constitutional symptoms (the oldest presented with leg ulcers at 59 years old), and 13 had severe PAN (digital necrosis, visceral organ injury), which was fatal in 2 cases at 9 m and 31 years of age. Of ten skin biopsies, four showed necrotizing arterial vasculitis and six showed nonspecific leukocytoclastic vasculitis or panniculitis. Among the 19 Israeli patients, all homozygous for the G47R mutation, 18 had cPAN, mainly livedo reticularis; 10 also had visceral PAN, diagnosed by 1 year of age in 6 cases, with stenosis and aneurysms of abdominal arteries, or renal cortical infarcts and hypertension. Neurologic involvement in 8 of these 19 patients was primarily peripheral, but in some cases, brain neuroimaging showed infarcts and ventricular hemorrhage, even in patients who lacked a clinical diagnosis of stroke. The four German siblings and the Turkish patient were compound heterozygotes for four different missense mutations. Of note, 10 of 13 patients with severe PAN had sustained benefit from treatment with anti-TNF agents.
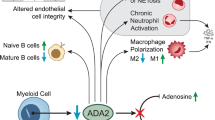
As mice lack a CECR1 orthologue, the NIH investigators used the zebrafish as a model, showing that transient knockdown of the cecr1b gene (orthologous to human CECR1) caused intracranial bleeding and marked neutropenia in embryos, which were prevented by providing wild-type, but not mutant, human CECR1 mRNA [2••]. Neither CECR1 mRNA nor ADA2 protein could be detected in normal human endothelial cells, suggesting that endothelial damage and vasculopathy in patients might involve macrophages, which secrete ADA2 and play a role in inflammation. Patient-derived monocytes showed normal GM-CSF induced differentiation into M1 (pro-inflammatory) macrophages, but defective M-CSF induced differentiation of into M2 (anti-inflammatory) macrophages. Human dermal endothelial cells co-cultured with patient, but not control monocytes, formed abnormal layers. Together, these findings led to a hypothesis that ADA2 deficiency, by impairing M2 macrophage differentiation, leads to M1 polarization, compromising endothelial integrity and establishing a vicious cycle of vasculopathy and inflammation [2••].
Phenotypic Diversity
Reports describing ~ 125 DADA2 patients have appeared to date (key features are summarized in Table 1). Some patients were screened for CECR1 mutations because of clinical suspicion, e.g., a family with Sneddon syndrome, a non-inflammatory arteriopathy with onset of livedo reticularis in the second decade and stroke in early adulthood [18•]. In other cases, bi-allelic CECR1 mutations were found unexpectedly when WES was performed in patients with a different phenotype, e.g., a single patient with HHV8-negative (idiopathic) Castelman disease and another with monocytopenia and M. avium complex infection (MonoMAC syndrome), but without mutation in GATA2 [17•, 31].
The use of WES has led to the discovery of DADA2 in a growing number of patients who present with pure red cell aplasia (PRCA), sometimes occurring with other cytopenias, and with or without overt signs of inflammation or vasculopathy [14•, 28•, 34•]. DADA2 patients with PRCA differ from patients with Diamond Blackfan anemia (DBA), in which PRCA is associated with mutations in genes for ribosomal proteins or GATA1. About 70% of typical DBA patients have elevated levels of erythrocyte ADA1 activity, which is not found in PRCA due to DADA2. It is uncertain whether the bone marrow dysfunction associated with DADA2 represents an intrinsic defect or is secondary, possibly due to impaired interaction (perhaps mediated via secreted ADA2) between components of the hematopoietic microenvironment and macrophages or some other myeloid cell type.
Various immunological abnormalities, mainly of the B cell compartment, were observed in the original DADA2 patients. Persistent autoimmunity has been documented in a few patients [15•, 38]. Two reports have now described 11 patients with DADA2 who presented primarily with antibody deficiency and infections, compatible in several cases with a diagnosis of common variable immune deficiency (CVID) [30, 37••]. Median age of these patients at evaluation was 22 years, and it was 8 years at onset of symptoms (Table 1). It has been postulated that the defect in B cell maturation/differentiation might arise from aberrant communication between the innate and adaptive immune systems.
The presence of inflammatory CNS and skin involvement both in DADA2 and in the Aicardi Goutieres syndrome (due to mutations in SAMHD1) have led to the discovery of increased expression of interferon genes (“interferon signature”) in peripheral blood of several DADA2 patients; genome-wide overexpression of neutrophil genes was also found in some of these patients [19••, 27, 38]. These findings have led to the proposal that ADA2 may be a regulator of neutrophil activation, and that the vasculopathy in DADA2 patients may be an indirect effect of marked chronic neutrophil activation [19••]. It is unclear how enhanced neutrophil activation, or for that matter defective M2 macrophage differentiation, might result in selective bone marrow failure in some patients, but PAN in others.
Together, the reports summarized in Table 1 have expanded the phenotype and the number of pathogenic CECR1 mutations associated with DADA2; they also highlight the variability in disease expression and the lack of a clear relationship between CECR1 genotype and clinical phenotype. All clinically affected patients have been deficient in circulating ADA2 activity, even when only one disease causing CECR1 variant can be identified (i.e., by routine methods, but without a search for large deletions). As CECR1 is highly polymorphic, measurement of plasma ADA2 activity is also useful in excluding DADA2 when DNA sequencing reveals only one pathogenic mutation (alone or in combination with a variant of uncertain significance). Two caveats should be mentioned: some apparently healthy but CECR1-mutated and ADA2-deficient individuals have been discovered among first-degree relatives of clinically affected patients, raising difficult questions about the need for and timing of therapy. Conversely, although DADA2 has an autosomal recessive pattern of inheritance, a few carriers with half-normal plasma ADA2 activity have exhibited some clinical features associated with DADA2, raising the possibility of CECR1 haploinsufficiency, perhaps involving effects of modifier genes.
Therapy
Multiple therapeutic options have been described in case reports and a few case series, but the choice of treatment for an individual patient depends on the predominant symptoms and severity. Immunosuppressive agents have controlled inflammation in some patients, but response is usually transient. Anti-TNF agents were effective in 10 of 13 Israeli patients with severe PAN [3••], and in 12 patients treated at the NIH for a median of 10 months with no new strokes or other serious complications; cPAN improved in these patients, although livedo persisted in some [39]. Nine of ten symptomatic patients in a series from the UK required anti-TNF therapy [26•]. Steroids were ineffective in six Turkish patients with PAN, but two benefitted from anti-TNF therapy [23•]. At an international workshop on DADA2 held in Bethesda, MD, in November 2016, there seemed to be a consensus that anti-TNF agents are a preferred treatment, but as the course of the disease is variable over time, it is not yet clear whether their use will indefinitely prevent the occurrence of strokes or the emergence of PAN. There is also uncertainty about the role of anti-platelet and anticoagulant prophylaxis for stroke, with some indication that it might increase the risk for hemorrhagic events.
Sirolimus, which can reduce M1 macrophage differentiation and IL-6 production, was effective in controlling inflammatory features of DADA2 in one of two affected siblings who both had markedly elevated serum IL-6 levels [15•]. A patient with Castelman-like disease and high serum IL-6 levels responded to tocilizumab [17•].
PRCA and other kinds of hematologic dysfunction have generally not responded well to immunosuppressive agents, such as azathioprine, MMF, and cyclosporine (there is little information about anti-TNF therapy in such patients). As bone marrow-derived monocytes and macrophages are the main source of secreted ADA2, it was postulated early on that hematopoietic stem cell transplantation (HSCT) might be of benefit, although there was concern that the inflammation and vasculopathy associated with DADA2 might increase the risk for complications in the peri-transplant period, such as stroke or sinusoidal obstruction syndrome (SOS). HSCT has been performed to date in more than a dozen patients with severe hematologic or immunologic involvement, and has been successful in all cases, in spite of various complications [14•, 15•, 35•, 40••]. Restoration of normal plasma ADA2 activity has occurred in all the cases, as early as 2 weeks post transplant, and all disease manifestations appear to have resolved, with a median follow-up of 18 months. In some patients transplanted prior to the discovery of DADA2, benefit has persisted with follow-up of 5–10 years.
As ADA2 is found in plasma, the possibility of treating its deficiency with infusions of fresh frozen plasma (FFP) has been considered. However, the half-life of ADA2 after administration of FFP appears to be too short to make this practical [39]. PEGylated bovine ADA1 has been used for more than 20 years as replacement therapy for patients with SCID due to ADA1 deficiency, and effective gene therapy for that disorder is now available. The possibility of developing analogous therapies for DADA2 is under investigation.
The Physiological Function(s) of ADA2
The function of ADA2 in humans and the pathogenesis of DADA2 are not understood, but are the subject of two main avenues of speculation. One hypothesis assumes that an important function of extracellular ADA2 must be to degrade extracellular Ado and thereby to control signaling through one or more of the four known G protein-coupled Ado receptors (A1, A2A, A2B, A3). Loss of ADA2 might be expected to result in elevated levels of Ado in plasma, possibly derived from excessive ATP breakdown at sites of inflammation or ischemia. The second hypothesis is that ADA2 is a growth or differentiation factor that acts by binding to unidentified cell-associated receptors to regulate the inflammatory process.
There is no evidence that loss of ADA2 alters the level of extracellular Ado. The concentrations of Ado and dAdo were normal (< 0.4 μM) in plasma of a patient with DADA2 (and in several others we have studied) [15•]. By contrast, in spite of having normal plasma ADA2 activity, these levels approach or exceed 10 μM in patients with SCID due to ADA1 deficiency [41]. These findings are consistent with the substrate affinities of ADA1 and ADA2, and the role of nucleoside transport in Ado metabolism. Thus, human ADA2 has very low affinity for Ado (and dAdo), with a Michaelis constant (Km) of ~ 2 mM, ~ 10,000-fold above the normal concentration of Ado in plasma and extracellular fluid (~ 0.2 μM). Cells possess bidirectional nucleoside transporters, which have higher affinity for Ado than ADA2 and function to rapidly equilibrate intra- and extracellular Ado concentrations. The concentration of free Ado in cells is kept vanishingly low by the actions of intracellular ADA1, which is a very efficient enzyme with a Km for Ado of ~ 20 μM, and Ado kinase (Km ~ 1 μM). As a result, any Ado generated in the extracellular space can very rapidly be taken up by erythrocytes or other nearby cells, where it is efficiently metabolized. Even without invoking any effect of so-called ecto-ADA (extracellular ADA1 bound to the glycoprotein CD26 present on many epithelial cells), these findings all support the conclusion that intracellular ADA1 plays a much greater role than extracellular ADA2 in controlling extracellular Ado. This must be true in mice, which lack both ADA2 and CD26-bound ADA1 [42]. Nevertheless, if one still believes that elevated extracellular Ado plays a significant role in the pathogenesis of DADA2, it might be worth considering a trial of PEGylated ADA1, which works by degrading Ado and dAdo in plasma after parenteral administration and is approved for treating SCID due to ADA1 deficiency.
That ADA1 and ADA2 are not functionally redundant is evident from the very different metabolic and clinical consequences associated with their deficiencies. This makes the hypothesis that secreted ADA2 is a growth or differentiation factor-attractive, but it is useful to appreciate that this notion arose from a fundamental misinterpretation of experimental findings. It derives from the isolation 20 years ago of a secreted “Insect Derived Growth Factor (IDGF),” renamed “ADAGF” after it was found to be a very active ADA, with a Km comparable to that of “mammalian cytoplasmic ADA,” i.e., ADA1 [43, 44]. The “growth stimulation” attributed to ADAGF was totally dependent on its ADA activity, and the phenomenon was shown more accurately to be a relief of growth inhibition caused by exogenous Ado, which was present at toxic concentrations in fly cell culture medium; the same apparent effect on cell growth could be achieved by adding bovine ADA1 [44, 45]. CECR1 was identified as the human homolog of IDGF/ADAGF [46], and ADA2 was shown to be its product, albeit far less enzymatically active than invertebrate ADAGFs [7, 47].
The hypothesis that secreted human ADA2 is a growth or differentiation factor has not been investigated in vivo, as a suitable animal model is lacking. However, Zavialov and his colleagues have reported that both exogenously added human ADA2 and ADA1, independent of enzymatic activity, could promote the division of cultured human T cells, and that ADA2, but not ADA1, could stimulate the T cell-dependent differentiation of monocytes into macrophages. These in vitro findings were highly dependent on the concentrations of ADA1 and ADA2 employed, and on other experimental conditions [6]. Zavialov’s laboratory has shown that ADA2 binds avidly to heparin sulfate and proteoglycans present on many cell types, although the consequences of this interaction are unknown [7]. More recently, these investigators have studied the ability of ADA2 to bind to various cells involved in immunity or inflammation [48]. A critical discussion of these experimental findings and their potential physiological relevance is beyond the scope of this review.
Conclusions
Almost 4 years after the first reports of its existence, DADA2 has entered the differential diagnosis of several serious conditions of interest to both pediatric and adult rheumatologists, including PAN and other forms of vasculitis, the autoinflammatory disorders, and disorders characterized by immune deficiency and dysregulation. In a subset of DADA2 patients, bone marrow failure dominates the clinical picture. Fortunately, many patients respond well to anti-TNF agents, which are employed by rheumatologists to treat RA and other diseases characterized by systemic inflammation. Because the manifestations of DADA2 are so varied, and the complications of vasculopathy potentially catastrophic, it behooves rheumatologists to appreciate the ways in which DADA2 may present, and to have a high index of suspicion, particularly in patients with compatible features who lack typical biomarkers for other conditions under consideration, or do not respond satisfactorily to immunosuppressive therapies. The diagnosis of DADA2 can be made or excluded not only by CECR1 gene sequencing but also by a relatively straightforward enzyme assay.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. 2017;18(8):832–42. https://doi.org/10.1038/ni.3777.
•• Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20. https://doi.org/10.1056/NEJMoa1307361. The first report of DADA2 as a cause of early-onset stroke. Provides experimental evidence supporting causalityof ADA2 deficiency and evidence implicating M1 macrophage polarization in pathogenesis
•• Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31. https://doi.org/10.1056/NEJMoa1307362. The first report of DADA2 as a cause of familial and sporadic PAN. Provides evidence of the efficacy of anti-TNF therapy in controlling autoinflammation
Schrader WP, Pollara B, Meuwissen HJ. Characterization of the residual adenosine deaminating activity in the spleen of a patient with combined immunodeficiency disease and adenosine deaminase deficiency. Proc Natl Acad Sci U S A. 1978;75(1):446–50.
Iwaki-Egawa S, Yamamoto T, Watanabe Y. Human plasma adenosine deaminase 2 is secreted by activated monocytes. Biol Chem. 2006;387(3):319–21. https://doi.org/10.1515/BC.2006.042.
Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. 2010;88(2):279–90. https://doi.org/10.1189/jlb.1109764.
Zavialov AV, Engstrom A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem J. 2005;391(Pt 1):51–7. https://doi.org/10.1042/BJ20050683.
Zavialov AV, Yu X, Spillmann D, Lauvau G, Zavialov AV. Structural basis for the growth factor activity of human adenosine deaminase ADA2. J Biol Chem. 2010;285(16):12367–77. https://doi.org/10.1074/jbc.M109.083527.
Poursharifi P, Saghiri R, Ebrahimi-Rad M, Nazem H, Pourpak Z, Moin M, et al. Adenosine deaminase in patients with primary immunodeficiency syndromes: the analysis of serum ADA1 and ADA2 activities. Clin Biochem. 2009;42(13–14):1438–43. https://doi.org/10.1016/j.clinbiochem.2008.10.019.
Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. 2016;14(1):51. https://doi.org/10.1186/s12969-016-0111-7.
Berkun Y, Segel R, Navon-Elkan P. Adenosine deaminase 2 deficiency: more than monogenic vasculitis. Isr Med Assoc J. 2017;19(7):435–7.
• Ozen S. The changing face of polyarteritis nodosa and necrotizing vasculitis. Nat Rev Rheumatol. 2017;13(6):381–6. https://doi.org/10.1038/nrrheum.2017.68. A thoughtful and informative recent review of PAN
Garg N, Kasapcopur O, Foster J 2nd, Barut K, Tekin A, Kizilkilic O, et al. Novel adenosine deaminase 2 mutations in a child with a fatal vasculopathy. Eur J Pediatr. 2014;173(6):827–30. https://doi.org/10.1007/s00431-014-2320-8.
• van Montfrans J, Zavialov A, Zhou Q. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):478. https://doi.org/10.1056/NEJMc1405506#SA1. HSCT from a matched allogeneic donor was performed for PRCA in a patient later discovered to have DADA2. Serum ADA2 and clinical status were normal 11 years post transplant
• Van Eyck L Jr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. 2015;135(1):283–7 e5. https://doi.org/10.1016/j.jaci.2014.10.010. Describes the complicated course and successful HSCT in one of two siblings with DADA2, and documents elevated IL-6 in both siblings
Van Eyck L, Liston A, Meyts I. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):478–9. https://doi.org/10.1056/NEJMc1405506#SA2.
• Van Eyck L, Liston A, Wouters C. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):480. https://doi.org/10.1056/NEJMc1405506#SA4. DADA2 as a cause of HHV8-negativer Castleman disease, effectively treated with anti-IL6
• Bras J, Guerreiro R, Santo GC. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):478–80. https://doi.org/10.1056/NEJMc1405506#SA3. DADA2 as a cause of familial Sneddon syndrome (onset of livedo racemosa and stroke in the second decade of life)
•• Belot A, Wassmer E, Twilt M, Lega JC, Zeef LA, Oojageer A, et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatr Rheumatol Online J. 2014;12:44. https://doi.org/10.1186/1546-0096-12-44. Disconvery of interfereon signature and overexpression of neutrophil genes in two patients with DADA2. Suggests inflammatory features and PAN may result from chronic neutophil activation
Gonzalez Santiago TM, Zavialov A, Saarela J, Seppanen M, Reed AM, Abraham RS, et al. Dermatologic features of ADA2 deficiency in cutaneous polyarteritis nodosa. JAMA Dermatol. 2015;151(11):1230–4. https://doi.org/10.1001/jamadermatol.2015.1635.
Abraham RS, Gibson LE. Error in complementary DNA nomenclature in “Dermatologic Features of ADA2 Deficiency in Cutaneous Polyarteritis Nodosa”. JAMA Dermatol. 2016;152(9):1065. https://doi.org/10.1001/jamadermatol.2016.2595.
Westendorp WF, Nederkoorn PJ, Aksentijevich I, Hak AE, Lichtenbelt KD, Braun KP. Unexplained early-onset lacunar stroke and inflammatory skin lesions: consider ADA2 deficiency. Neurology. 2015;84(20):2092–3. https://doi.org/10.1212/WNL.0000000000001581.
• Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A case series of adenosine deaminase 2-deficient patients emphasizing treatment and genotype-phenotype correlations. J Rheumatol. 2015;42(8):1532–4. https://doi.org/10.3899/jrheum.150024. Highlights weak correlation between genotype and phenotype
Fellmann F, Angelini F, Wassenberg J, Perreau M, Arenas Ramirez N, Simon G, et al. IL-17 receptor A and adenosine deaminase 2 deficiency in siblings with recurrent infections and chronic inflammation. J Allergy Clin Immunol. 2016;137(4):1189–96 e1-2. https://doi.org/10.1016/j.jaci.2015.07.053.
Keer N, Hershfield M, Caskey T, Unizony S. Novel compound heterozygous variants in CECR1 gene associated with childhood onset polyarteritis nodosa and deficiency of ADA2. Rheumatology (Oxford). 2016;55(6):1145–7. https://doi.org/10.1093/rheumatology/kew050.
• Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of adenosine deaminase type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheumatol. 2016;68(9):2314–22. https://doi.org/10.1002/art.39699. Highlights phenotypic heterogeneity in related patients homozygous for the same CECR1 mutation
Uettwiller F, Sarrabay G, Rodero MP, Rice GI, Lagrue E, Marot Y, et al. ADA2 deficiency: case report of a new phenotype and novel mutation in two sisters. RMD Open. 2016;2(1):e000236. https://doi.org/10.1136/rmdopen-2015-000236.
• Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. 2016;177:316–20. https://doi.org/10.1016/j.jpeds.2016.06.058. Highlights PRCA and other hematologic disorders in patients with DADA2 from several Arab families
• Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (Oxford). 2016;55(5):902–10. https://doi.org/10.1093/rheumatology/kev439. Highlights phenotypic heterogeneity in related patients homozygous for the same CECR1 mutation
Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. 2016;36(3):179–86. https://doi.org/10.1007/s10875-016-0245-x.
• Hsu AP, West RR, Calvo KR, Cuellar-Rodriguez J, Parta M, Kelly SJ, et al. Adenosine deaminase type 2 deficiency masquerading as GATA2 deficiency: successful hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2016;138(2):628–30 e2. https://doi.org/10.1016/j.jaci.2016.03.016. DADA2 as a cause of MonoMAC syndrome, successfully treated by HSCT
Poswar Fde O, da Fonseca RM, de Albuquerque LC, Zhou Q, Jardim LB, Monte TL, et al. Adenosine deaminase 2 deficiency presenting as spastic paraplegia and systemic vasculitis. J Neurol. 2016;263(4):818–20. https://doi.org/10.1007/s00415-016-8070-y.
Elbracht M, Mull M, Wagner N, Kuhl C, Abicht A, Kurth I, et al. Stroke as initial manifestation of adenosine deaminase 2 deficiency. Neuropediatrics. 2017;48(2):111–4. https://doi.org/10.1055/s-0036-1597611.
• Hashem H, Egler R, Dalal J. Refractory pure red cell aplasia manifesting as deficiency of adenosine deaminase 2. J Pediatr Hematol Oncol. 2017; https://doi.org/10.1097/MPH.0000000000000805. Successful HSCT for PRCA following diagnosis of DADA2
• Hashem H, Vatsayan A, Gupta A, Nagle K, Hershfield M, Dalal J. Successful reduced intensity hematopoietic cell transplant in a patient with deficiency of adenosine deaminase 2. Bone Marrow Transplant. 2017; https://doi.org/10.1038/bmt.2017.173.
Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017; https://doi.org/10.1136/annrheumdis-2016-210802.
•• Schepp J, Proietti M, Frede N, Buchta M, Hubscher K, Rojas Restrepo J, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol. 2017;69(8):1689–700. https://doi.org/10.1002/art.40147. Screening idenitfied DADA2 in 11 (6%) of 181 patients with antibody deficiency (CVID)
Skrabl-Baumgartner A, Plecko B, Konig N, Hershfield M, Gruber-Sedlmayr U, Lee-Kirsch MA. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol. 2017;15(1):67. https://doi.org/10.1186/s12969-017-0193-x.
Ombrello A, Stone D, Hoffmann P, Jones A, Barham B, Barron K et al. The deficiency of adenosine deaminase type 2—results of therapeutic intervention. Pediatric Rheumatology. 2015;13 (Suppl 1). doi:https://doi.org/10.1186/1546-0096-13-S1-O40.
•• Hashem H, R A, Kumar AR, Müller I, Babor F, Bredius R et al. Hematopoietic stem cell transplantation rescues the hematological, immunological and vascular phenotype in DADA2. Blood. 2017, in Press. Reviews the experience with HSCT performed in 14 patients with DADA2.
Azzari C, la Marca G, Resti M. Neonatal screening for severe combined immunodeficiency caused by an adenosine deaminase defect: a reliable and inexpensive method using tandem mass spectrometry. J Allergy Clin Immunol. 2011;127(6):1394–9. https://doi.org/10.1016/j.jaci.2011.03.040.
Richard E, Arredondo-Vega FX, Santisteban I, Kelly SJ, Patel DD, Hershfield MS. The binding site of human adenosine deaminase for CD26/dipeptidyl peptidase IV: the Arg142Gln mutation impairs binding to cd26 but does not cause immune deficiency. J Exp Med. 2000;192(9):1223–36.
Homma K, Matsushita T, Natori S. Purification, characterization, and cDNA cloning of a novel growth factor from the conditioned medium of NIH-Sape-4, an embryonic cell line of Sarcophaga peregrina (flesh fly). J Biol Chem. 1996;271(23):13770–5.
Homma KJ, Tanaka Y, Matsushita T, Yokoyama K, Matsui H, Natori S. Adenosine deaminase activity of insect-derived growth factor is essential for its growth factor activity. J Biol Chem. 2001;276(47):43761–6. https://doi.org/10.1074/jbc.M105088200.
Zurovec M, Dolezal T, Gazi M, Pavlova E, Bryant PJ. Adenosine deaminase-related growth factors stimulate cell proliferation in Drosophila by depleting extracellular adenosine. Proc Natl Acad Sci U S A. 2002;99(7):4403–8. https://doi.org/10.1073/pnas.062059699.
Riazi MA, Brinkman-Mills P, Nguyen T, Pan H, Phan S, Ying F, et al. The human homolog of insect-derived growth factor, CECR1, is a candidate gene for features of cat eye syndrome. Genomics. 2000;64(3):277–85. https://doi.org/10.1006/geno.1999.6099.
Charlab R, Valenzuela JG, Andersen J, Ribeiro JM. The invertebrate growth factor/CECR1 subfamily of adenosine deaminase proteins. Gene. 2001;267(1):13–22.
Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. 2017;74(3):555–70. https://doi.org/10.1007/s00018-016-2357-0.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Michael S Hershfield receives grant support from Leadiant Biosciences, Inc., the manufacturer of Adagen, a PEGylated adenosine deaminase used to treat patients with ADA1 deficiency.
Hasan Hashem, Susan J Kelly, and Nancy J Ganson declare they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Orphan Diseases
Rights and permissions
About this article

Cite this article
Hashem, H., Kelly, S.J., Ganson, N.J. et al. Deficiency of Adenosine Deaminase 2 (DADA2), an Inherited Cause of Polyarteritis Nodosa and a Mimic of Other Systemic Rheumatologic Disorders. Curr Rheumatol Rep 19, 70 (2017). https://doi.org/10.1007/s11926-017-0699-8
Published:
DOI: https://doi.org/10.1007/s11926-017-0699-8