
Abstract
Purpose of Review
Laboratory criteria for the classification of antiphospholipid syndrome include the detection of a lupus anticoagulant and/or anticardiolipin and anti-β2-glycoprotein I antibodies. However, the majority of patients who test positive in these assays do not have thrombosis. Current risk-stratification tools are largely limited to the antiphospholipid antibody profile and traditional thrombotic risk factors.
Recent Findings
Novel biomarkers that correlate with disease activity and potentially provide insight into future clinical events include domain 1 specific anti-β2GPI antibodies, antibodies to other phospholipids or phospholipid/protein antigens (such as anti-PS/PT), and functional/biological assays such as thrombin generation, complement activation, levels of circulating microparticles, and annexin A5 resistance. Clinical risk scores may also have value in predicting clinical events.
Summary
Biomarkers that predict thrombosis risk in patients with antiphospholipid antibodies have been long sought, and several biomarkers have been proposed. Ultimately, integration of biomarkers with established assays and clinical characteristics may offer the best chance of identifying patients at highest risk of APS-related complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The antiphospholipid syndrome (APS) is one of the most common acquired thrombophilias, and is characterized by recurrent thrombosis and/or obstetrical morbidity in the presence of antiphospholipid antibodies (aPL), specifically lupus anticoagulant (LA), anti-β2-glycoprotein I (anti-β2GPI), and/or anti-cardiolipin (aCL) antibodies [1•]. Thrombi occur most commonly in the deep veins of the lower extremities and the cerebral arterial circulation [2]; however, patients may develop thromboses in more unusual locations such as the hepatic veins, visceral veins, or the cerebral venous circulation. Obstetrical criteria for APS include one or more miscarriages at or beyond the 10th week of gestation, severe pre-eclampsia or eclampsia causing premature birth of one or more morphologically normal neonates before the 34th week of gestation, and/or three or more consecutive, unexplained, spontaneous abortions before the 10th week of gestation (Table 1). Rare patients (<1%) develop catastrophic antiphospholipid syndrome (CAPS) [3, 4], which is diagnosed by the presence of small vessel thrombosis in three or more organs within a period of 1 week in the presence of aPL, and is associated with a high mortality rate (~50%) [1•, 4]. Other manifestations commonly seen in patients with aPL, such as thrombocytopenia, livedo reticularis, skin ulcers, transient ischemic attacks, seizures, and migraine [5, 6] are not included in the diagnostic classification, but should alert physicians to the possibility of APS, especially in patients who also have thrombosis or pregnancy loss.
Pathogenesis of APS
Interactions with Coagulation-Related Proteins and Inhibitors
One of most frequently identified prothrombotic mechanisms of aPL is inhibition of natural anticoagulant activities. APL have been reported to inhibit the activation of protein C [7,8,9,10] as well as the ability of active protein C to inactivate factors V and VIII [11, 12]. These activities are mediated by antibodies to β2GPI and/or prothrombin [13,14,15,16], and may require the presence of phosphatidylethanolamine [17]. In addition, aPL inhibit heparin binding and activation of antithrombin [18], as well as the activity of the tissue factor pathway inhibitor [19]. Antiphospholipid antibodies may also inhibit fibrinolysis, at least in part by neutralizing the ability of β2GPI to stimulate the activity of tissue-type plasminogen activator [20]. Finally, aPL may block the anticoagulant activity of annexin A5 by impairing its ability to form a lattice on procoagulant anionic phospholipids in a β2GPI-dependent manner [21, 22].
Activation of Vascular Cells
There is general consensus that aPL activate vascular cells, a property thought to contribute significantly to the pathogenesis of APS [23, 24]. aPL activate endothelial cells in a β2GPI-dependent manner [25,26,27]; activation of endothelial cells leads to disruption of the normally anticoagulant endothelial surface and transformation to a prothrombotic phenotype. Endothelial cells activated by aPL demonstrate increased expression of cell adhesion molecules (E-selectin, VCAM-1, ICAM-1) and tissue factor [25, 26, 28], and decreased elaboration of endothelial cell-derived nitric oxide [29]. The pathways and mechanism of cellular activation are not completely defined, and several receptor-mediated pathways have been suggested involving annexin A2, TLR4/NF-ĸB, LRP-8, TLR2, and TLR7, among others [30, 31, 32•]. Mice deficient in annexin A2, TLR4, or LRP-8, as well as those treated with an NF-ĸB inhibitor, are relatively protected from the enhanced thrombosis that occurs following passive infusion of aPL [33,34,35,36]. In addition to endothelial cells, monocytes are also activated by aPL in the presence of β2GPI; this occurs in lipid raft structures via annexin A2-mediated mechanisms [37], though recent studies have suggested, as with endothelial cells, important roles for several members of the TLR family including TLR2.
Though direct binding of β2GPI to unstimulated platelets has not been well characterized, platelets are activated in the presence of aPL/anti-β2GPI antibodies. In a non-flow system, aPL activate platelets in the presence of subthreshold concentrations of thrombin in a p38 MAP-kinase-dependent manner [38], while under flow, aPL enhance adhesion of platelets to collagen through a process dependent on platelet glycoprotein 1b and apoER2 [39, 40]. Several studies have also demonstrated that aPL interact with placental trophoblasts, leading to an inflammatory response that may underlie the pathogenesis of aPL-associated fetal loss [41].
Complement Activation
The role of complement activation in APS was first demonstrated in murine models of aPL-associated pregnancy loss [42, 43]. Complement products C3a and C5a were found to cause placental inflammation, and mice deficient in C3, C4, C5, or the C5a receptor were protected from fetal loss induced by passive infusion of aPL IgG [44]. Since then, it has been demonstrated that complement activation contributes to aPL-mediated thrombosis in mice as demonstrated by the ability of C5 inhibition to prevent thrombosis in animals receiving passive infusion of anti-β2GPI antibodies [45, 46, 47••]. Complement activation by aPL also generates the potent inflammatory mediator C5a, which recruits monocytes and neutrophils, activates endothelial cells, and induces expression of tissue factor [48, 49]. There is some evidence supporting activation of both the classical and alternative complement pathways in patients with catastrophic APS [50•], and several case reports document the successful use of eculizumab (humanized anti-C5a monoclonal antibody) in CAPS and in patients with APS complicating renal transplantation [51•, 52, 53••, 54, 55].
Diagnosis of APS
The classification of “definite APS” is based on the Sapporo criteria, which were first proposed in 1999 [56] and updated in 2006 [1•]. These include clinical and laboratory criteria (Table 1), and at least one of each must be present to make a diagnosis. Since the clinical criteria, thrombosis and pregnancy loss, are relatively prevalent in the general population and have many causes, laboratory investigations are central to the diagnosis of APS. These include the presence of a persistently positive lupus anticoagulant detected according to ISTH guidelines, and/or positive anticardiolipin (aCL) antibodies (IgG or IgM) exceeding 40 IgG or IgM antiphospholipid units, and/or anti-β2GPI antibodies (IgG or IgM) at levels exceeding the 99th percenle in an enzyme-linked immunosorbent assay. To improve specificity, at least two assays should be performed to evaluate for each of the four ISTH criteria for detecting LA [57]. To minimize the risk of establishing a diagnosis based on transient aPL, recommendations suggest performing assays twice, with samples obtained at least 12 weeks apart [57, 58]. It is important to recognize that these criteria were initially proposed to standardize inclusion of patients into clinical studies. While they are widely applied as diagnostic tools, they were designed primarily for classification and have several shortcomings in clinical practice. For example, they do not account for patients who have persistent LA and/or aPL but have only non-criteria manifestations of APS, or for patients who have clinical criteria for APS but have only low to moderate titers of IgG/IgM aCL and anti-β2GPI. Occasional patients with clinical manifestations of APS lack positivity in any of the standard diagnostic laboratory studies, and are sometimes termed to have “seronegative APS” (Fig. 1), though the specificity of this term is uncertain. Some of these patients may have IgA antibodies against aCL or β2GPI [59, 60•, 61, 62, 63•], or antibodies against other antigens such as phosphatidylserine, phosphatidylethanolamine, prothrombin, annexin A2 [64, 65], annexin A5 [66], or vimentin/cardiolipin complexes [62, 67].

Antiphospholipid antibodies and APS-related events: A proportion of patients with thrombosis and pregnancy morbidity meeting clinical criteria for APS are positive for aPL by standard diagnostic criteria and are diagnosed with definite APS. Other patients with clinical criteria of APS are negative by standard laboratory criteria, termed “seronegative APS.” A large proportion of individuals with persistent aPL represent asymptomatic carriers, or individuals with “pre-APS” who may or may not develop APS-related clinical events in the future. Biomarkers of disease activity may have the greatest utility in the latter two categories
Thrombotic Risk Assessment in APS
The current classification criteria for APS (and aPL) provide relatively little information about the risk of recurrent thromboembolic events in an individual patient. This is an important clinical issue, since current guidelines recommend indefinite anticoagulation for patients with APS. Even more difficult is predicting the risk of thrombosis or obstetric morbidity in an individual with asymptomatic aPL, which may occur in a few percent of healthy individuals without a history of thrombotic events and in as many as 11% to 86% of individuals with systemic lupus erythematosus (SLE) [68].
The aPL profile, which refers to the type (LA, aCL, anti-β2GPI) and number of aPL (single, double, or triple positive), is the most extensively studied and validated risk stratification strategy in patients with aPL; however, it is a relatively insensitive marker for predicting thrombosis, its prognostic value is suboptimal, it may not consistently identify patients at greatest risk for obstetric morbidity, and it is likely to be significantly affected by poorly characterized factors underlying the lack of concordance in results of aPL assays performed in different clinical laboratories. Moreover, the aPL profile cannot be used to assess response to therapy or to identify patients with APS who may safely be treated with a shorter duration of anticoagulation. However, recent insights into the pathogenic mechanisms underlying APS have led to the emergence of novel biomarkers that reflect disease activity and cellular activation.
Antiphospholipid Antibody Profile
The criteria tests for aPL (LA, aCL, and anti-β2GPI) detect antibodies with overlapping, but not identical, specificities. Several retrospective and prospective studies have demonstrated that LA positivity is the strongest risk factor for both arterial and venous thrombosis in patients with and without SLE [69]. There is significant variation in strength of association in different studies that may reflect different methods used to detect LA, or the variable inclusion of LA that were not persistently positive. Retrospective and prospective studies have not shown a consistent association between aCL and thrombosis [70, 71]. In a systematic review of 25 observational studies including over 7000 patients, Galli et al. demonstrated that LA were associated with both venous (OR range, 4.09–16.2) and arterial (OR range, 8.65–10.84) thrombotic events; however, aCL were associated with thrombotic events in less than half of the studies [72]. In the Leiden Thrombophilia Study, LA was associated with a higher risk of thrombosis (OR 3.6, 95% CI 1.2–10.9) than anti-β2GPI (OR 2.4, 95% CI 1.3–4.2) and antiprothrombin (anti-PT) antibodies (OR 1.4, 95% CI 1.0–2.1) [73]. Over the past decade, β2GPI has been identified as a key antigen in APS, which has led to a focus on anti-β2GPI as the more clinically significant and predictive aPL. Consistent with this hypothesis, several retrospective studies showed that anti-β2GPI antibodies indeed correlate with thrombotic risk [73,74,75]; however, recent studies suggest that the thrombotic risk conferred by anti-β2GPI antibodies may be more modest, with odds ratios between 1.5 and 2.5 [74]. More recent data, discussed below, suggests that epitope specific domain1 anti-β2GPI antibodies may be more predictive of clinical events. In the systematic review by Galli et al. that included 28 studies with 4394 patients and 1973 controls, only 57% of associations of anti-β2GPI antibodies with thrombosis were significant [72]. The proportion of significant associations increased to 71% when only studies of patients with SLE were considered [72]. However, most of these studies were retrospective, used different methods of measuring aPL, and did not control for other thrombotic risk factors. In the prospective Warfarin in APS (WAPS) study that included 462 patients with persistent LA, IgG anti-β2GPI antibodies were associated with both arterial and venous thrombosis [70]. A meta-analysis of 25 studies examining the association of different aPL with recurrent pregnancy loss demonstrated that LA was most strongly associated with late recurrent pregnancy loss (OR 7.79, 95% CI 2.30–26.45) followed by IgG aCL (OR 3.57, 95% CI 2.26–5.65) and IgM aCL (OR 5.61, 95% CI 1.26–25.03) [76]. This meta-analysis did not comment on the association of anti-β2GPI antibodies with recurrent pregnancy loss due to a lack of methodologically consistent studies evaluating this outcome [76]. Although LA is the single test most predictive of the thrombotic phenotype, current assays for LA such as the DRVVT, while sensitive, are difficult to quantify given the absence of a suitable standard of activity. Thus, LA may be false positive, and a weak LA alone may be an epiphenomenon rather than causative, especially when interpreted in light of the fact that thrombosis is a common occurrence in the general population. Another study has demonstrated that lupus anticoagulants that are dependent upon the presence of β2GPI for their activity may correlate more strongly with a history of thrombosis (OR 42.3; 95% CI 194.3–9.9) than β2GPI-independent LA [77]. These studies require confirmation.
Over the past decade, numerous studies have shown that the risk of thrombosis increases with the number of positive tests for aPL in APS patients as well as individuals with asymptomatic persistent aPL. For example, in the WAPS study, there was a significantly increased risk of thrombosis in patients with both LA and anti-β2GPI antibodies (OR 4.1, 95% CI 1.3–13.5) [70]. In The Leiden Thrombophilia Study, LA positivity along with either anti-β2GPI or antiprothrombin antibodies was associated with a significantly increased risk of thrombosis compared to LA alone (OR 10.1, 95% 1.3–79.8) [73]. Pengo et al. reported a cumulative incidence of recurrent thrombosis of 12.2, 26.1, and 44.2% after 1, 5, and 10 years of follow-up in a retrospective analysis of 160 APS patients positive for LA, aCL, and anti-β2GPI—so-called “triple positive” patients, 123 of whom were on long-term anticoagulation [78]. In a prospective study of 104 triple positive aPL carriers, the rate of thromboembolism was 5.3% per year with a cumulative incidence rate of 37.1% over 10 years [79]. Other retrospective and prospective studies have confirmed the association of triple positivity with thrombosis in adults with APS (OR 5.24, 95% CI 1.5–18.3) [80] and asymptomatic aPL carriers [81•, 82•]. Triple positivity for LA, aCL, and anti-β2GPI has also been associated with history of late pregnancy loss (OR 16.2, 95% CI 0.9–292) and unsuccessful subsequent pregnancy (OR 34.4, 95% CI 3.5–335.1) [83]. Based on these observations, the 2006 revision of the Sapporo criteria recommended that patients should be classified as those with only one positive aPL and those with two or three positive aPL [1•].
There is minimal debate regarding the association of clinical manifestations of APS with LA, IgG, and IgM isotypes of aCL and anti-β2GPI antibodies that are included in the diagnostic criteria; however, the clinical importance of isolated IgA aCL and anti-β2GPI antibodies remains controversial. IgA aPL have been shown to be thrombogenic in murine experiments [84]. Previous studies have also highlighted the high prevalence of IgA anti-β2GPI antibodies in individuals with SLE, particularly in Afro-Caribbean populations [85, 86]. Others have reported an association between IgA anti-β2GPI and thromboembo-lic events, especially in patients with SLE [87, 88]. These antibodies usually occur in combination with other isotypes of anti-β2GPI making it difficult to evaluate their independent contribution to thrombotic risk. There are several case reports of patients who meet clinical criteria for APS that are positive for IgA anti-β2GPI antibodies in the absence of IgG or IgM antibodies (“seronegative-APS”). In the absence of standardized assays and well-designed prospective studies, we cannot recommend testing for IgA aPL in all patients with clinical manifestations of APS. IgA aPL testing may be useful in patients with "seronegative APS", especially those with SLE.
“Non-criteria” Antiphospholipid Antibodies
A large number of aPL directed against a variety of phospholipid-binding proteins have been identified [89]. The most promising of these in thrombotic APS recognize two major phospholipid-binding antigens—epitope specific (domain 1) anti-β2GP1 antibodies and antibodies to prothrombin (and phosphatidylserine/prothrombin complexes; Table 2). Others recognize vimentin/cardiolipin complexes, annexin A2 and annexin A5. The significance of these latter antibodies remains uncertain though it has been suggested that they may activate distinct intracellular signaling pathways leading to the pleomorphic manifestations of APS [90]. They may have particular relevance in the evaluation of patients who present with the classical clinical manifestations of APS with negative or subthreshold results on the standard diagnostic assays.
Anti-β2GPI-domain1 Antibodies
Antibodies to β2GPI can be directed against any of the five domains of β2GPI. DeLaat et al. demonstrated that IgG antibodies that recognize the Gly40-Arg43 epitope in the first domain of β2GPI, called anti-β2GPI-domain 1 antibodies, are associated with LA activity and are more strongly associated with a history of thrombosis and obstetrical morbidity compared to antibodies directed against other regions of the protein [91, 92]. A prospective study reported that IgG anti-β2GPI domain 1 antibodies were more often persistent at 12 weeks, associated with triple positivity, and correlated with thrombotic risk [93••]. In a recent study, anti-β2GPI-domain 1 antibodies predicted clinical events with an OR of 17 (95% CI, 7.1–40.5) although they did not add to the diagnostic accuracy of the standard aPL panel since anti-β2GPI antibodies were even more sensitive and almost as specific for patients with thrombosis [94]. However, this study also reported that β2GPI-domain 1 antibodies identified triple positive patients and those with thrombosis and β2GPI-dependent LA [94]. Mahler et al. detected anti-β2GPI domain 1 antibodies in 122/144 patients with APS and 1/200 (0.5%) of controls without APS yielding 85% sensitivity and 99.5% specificity [95]. Assays for anti-β2GP1-domain 1 antibodies might be particularly useful in identifying asymptomatic carriers with clinically significant anti-β2GP1 antibodies that may lead to clinical complications. A commercial assay for anti-β2GP1-domain 1 antibodies has been developed (Quanta Flash β2GPI-domain1, Inova Diagnostics); however, this is generally limited to the research setting.
Antiprothrombin and Antiphosphatidylserine/Prothrombin Antibodies
Anti-PT antibodies are detected in a 50–90% of LA-positive individuals [96]. To be antigenically recognized, prothrombin (PT) must either be coated on activated plates or combined with anionic phosphatidylserine (PS) to form PS/PT complexes. These antibodies are not associated with hypoprothrombinemia in the majority of cases [97]; however, in rare cases, LA-associated hypoprothrombinemia causes a significant bleeding diathesis [98]. Many anti-PT antibodies cause LA activity [97, 99,100,101]. Anti-PT and anti-PS/PT antibodies can co-exist and appear to represent distinct antibody populations [102]. The clinical significance of anti-PT antibodies, however, is still a matter of debate. While several studies have reported that anti-PT antibodies are associated with arterial or venous thrombosis [103,104,105], others have failed to demonstrate this association [106,107,108]. On the other hand, most studies evaluating the significance of aPS/PT antibodies have demonstrated an association with venous thrombosis [104, 105, 107,108,109,110,111]. Consistent with this, a systematic review of data from over 7000 individuals from 38 studies evaluating anti-PT and 10 studies evaluating anti-PS/PT as a marker of thrombosis noted that there was a stronger association of anti-PS/PT (OR 5.11, 95% CI 4.2–6.3) than of anti-PT with arterial or venous thrombotic events (OR 1.82, 95% CI 1.44–2.75) [112]. Of the seven studies that evaluated both anti-PS and anti-PS/PT, 90% identified an association of anti-PS/PT with thrombosis compared with only 45.5% that identified an association of anti-PS with thrombosis [112]. In another study evaluating 23 possible combinations of aPL specificities as a predictor of APS-related clinical events in 230 patients with SLE, a combination of LA, anti-PS/PT, and anti-β2GP1 had the best diagnostic accuracy for both thrombosis and pregnancy loss [113]. This has not yet been validated in patients with primary APS. Although well described, the association of aPT with thrombosis appears to be less strong than that of LA or anti-β2GP1 [114, 115]. While the current evidence is not enough to recommend routine testing for anti-PS/PT and anti-PT antibodies in patients with APS, this remains a promising area of investigation, and antibodies specific to PT, particularly anti-PS/PT, may prove useful risk stratification tools in APS.
Antiphosphatidylethanolamine Antibodies
Phosphatidylethanolamine (PE) is one of the primary lipid components of the cell membrane. While sera from APS patients usually react with negatively charged phospholipids and cofactors (e.g., β2GP1), sera reactive with PE, a neutral phospholipid, are less commonly observed. In several reports, aPE antibodies have been reported in patients with SLE and thrombosis in the absence of LA and aCL [116••, 117, 118], as well as in other patients with vasculopathy and livedo reticularis [119, 120]. In an analysis of 140 patients with thrombotic events and 136 controls, aPE was the only non-criteria aPL significantly more prevalent in patients than in controls (14.3 vs. 5.1%, P = 0.014) [121]. In another multicenter study, aPE were found in 15% of patients compared with 3% of controls [122]. Interestingly, 63% of the aPE-positive patients were negative for the standard serologic criteria for APS and the majority of them had venous thrombosis, half of which was recurrent VTE [122]. In contrast, a study by Bertolaccini et al. failed to demonstrate an association of aPE with thrombotic events in SLE [103]. Although plasma reactivity to PE is associated with LA activity and aCL, the direct relationship of aPE with LA, or with thrombotic mechanisms in APS, is not clear [123, 124]. Moreover, there have been no standards developed for standardization of anti-PE measurements, and their associations with clinical events have been reported by only a limited number of laboratories. While there is currently insufficient data to recommend testing for aPE in patients with APS, this might be considered in patients with seronegative APS.
Antibodies to Annexin A2 and Annexin A5
APS is associated with resistance to the anticoagulant effect of annexin A5 [125]. This property has been proposed to distinguish patients with APS from asymptomatic individuals with aPL as well as patients with venous thromboembolism but no evidence of aPL [126]. Anti-annexin A5 antibodies have been described in APS [127], and have been associated with placental thrombosis and fetal absorption in a mouse model. However, clinical studies have failed to consistently demonstrate a strong association with thrombosis [127] or pregnancy complications [128, 129]. Annexin A2 is implicated in aPL medicated cellular activation [26]. Although anti-annexin A2 antibodies have been described in APS, their clinical significance is uncertain.
Antibodies Against Vimentin/Cardiolipin Complexes
Vimentin is a ubiquitous cytoskeletal protein. In patients with SLE, antivimentin antibodies have been described that correlate with aCL [130]. Ortona et al. identified vimentin-cardiolipin complexes as an antigenic target in APS and demonstrated antivimentin/cardiolipin antibodies in 92.5% (37/40) of patients with APS and 55.2% (16/29) with seronegative APS [67]. Antivimentin/cardiolipin antibodies induced interleukin receptor-associated kinase phosphorylation and nuclear factor-κB activation in endothelial cells suggesting a pathophysiologic role. However, it is not clear that these antibodies are an APS-specific biomarker since they were present in 16.7% and 6.7% of subjects with rheumatoid arthritis and non-APS-related thrombosis, respectively.
Clinical Risk Scores
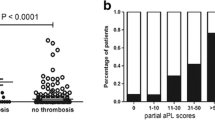
In addition to the aPL profile, investigators have developed several risk scores combining clinical and/or laboratory findings in an attempt to better identify individuals at risk of APS-related thrombosis. The antiphospholipid score (aPL-S) includes LA, aCL, and anti-β2GPI positivity and titers, and was developed to predict risk of APS-related clinical events in patients with autoimmune disorders [131] and subsequently validated in an independent cohort of patients with SLE [132]. The global APS score (GAPSS) was initially developed to predict both APS-related thrombosis and pregnancy loss in a cohort of patients with SLE [133]. In contrast to the aPL-S, the GAPSS included conventional cardiovascular risk factors in addition to aPL profiles; points assigned on the basis of a multivariable prediction model were 3 for hyperlipidemia, 1 for arterial hypertension, 5 for aCL IgG/IgM, 4 for anti-β2GPI IgG/IgM, 3 for antiphosphatidylserine/prothrombin IgG/IgM, and 4 for LA, and appeared to improve prediction of APS-related clinical events compared to aPL profile alone; a score GAPSS values ≥10 had the best diagnostic accuracy. The authors subsequently validated this score in a cohort of patients with primary APS [134•]. Independent validation of the GAPSS in a Japanese cohort of patients with autoimmune disease as well as primary APS also confirmed higher scores in patients with thrombosis, with maximum diagnostic accuracy for GAPSS >6; however, the predictive value of the GAPSS for pregnancy loss could not be validated [135]. Concerns about this scoring system include weighting aCL, for which the relationship to thrombosis is uncertain; greater than lupus anticoagulants; and the inclusion of antiphosphatidylserine/prothrombin IgG/IgM that are not routinely performed. Also, the optimal cutoff on the GAPSS score was different in all of these studies, which may be attributed to differences in the baseline characteristics of the cohort. While the GAPSS score may add to the utility of aPL in predicting thrombosis in APS, it still needs to be validated in patients with asymptomatic aPL and whether it will prove useful in clinical practice remains to be determined.
Other Risk Factors for APS-Related Clinical Events
While persistence and high levels of aPL, along with the aPL profile, are the major risk factors for thrombosis in APS, the presence of traditional risk factors such as inherited thrombophilia, systemic inflammatory disorders such as SLE, cancer, obesity, immobilization, smoking, pregnancy, the use of oral contraceptives, and a history of previous thrombosis also increase thrombotic risk [1•]. SLE, hypocomplementemia, decreased platelet counts, and a previous history of thrombosis and pregnancy failure are also additional risk factors for pregnancy failure [136]. The PROMISSE (Predictors of Pregnancy Outcome: Biomarkers in Antiphospholipid Antibody Syndrome and Systemic Lupus Erythematosus) study identified the presence of LA (OR 8.32, 95% CI 3.59–19.26), physician global assessment score >1 (OR 4.02, 95% CI 1.84–8.82), and low platelet count (OR 1.33, 95% CI 1.09–1.63 per 50 × 109/L) as predictors of adverse pregnancy outcomes [137]. Uterine Doppler ultrasound parameters can also identify women with SLE or APS at risk for obstetric complications [138, 139].
Emerging Biologic and Functional Biomarkers
Recognizing the limitations of current diagnostic and risk stratification tools in APS, there has been increasing interest in novel biomarkers based on recent insights into pathophysiologic mechanisms underlying APS that may correlate better with disease activity and could help in evaluating response to anticoagulation and other therapies.
The thrombin generation assay, a global coagulation assay that evaluates the generation of thrombin under in vitro conditions that attempt to approximate in vivo conditions, is one of the earliest markers to be evaluated as a measure of “clotting potential” [140, 141]. There has been limited evaluation of thrombin generation assays in APS; however, early studies demonstrated that anti-β2GPI antibodies with LA activity cause prolongation of the lag time similar to the prolongation in clotting times in the DRVVT and aPTT assays [142]. Moreover, in patients with LA, there is a marked inability of activated protein C to diminish peak thrombin generation indicating acquired resistance to protein C [143,144,145]. Devreese et al. demonstrated that the ratio of peak height (of thrombin generation) and lag time correlated reliably with LA activity detected in standard mixing tests [144]. They supplemented this approach with measurement of P-selectin and factor VII, markers of hypercoagulability, to develop a layered strategy with sensitivity and specificity for future thrombotic events [146]. More recently, Efthymiou et al. used thrombin generation assays to evaluate intensity of anticoagulation in thrombotic APS and non-APS patients [147••]. Endogenous thrombin potential and peak thrombin generation correlated inversely with INR; however, 20% of patients with APS had increased peak thrombin generation that exceeded the expected relative to the intensity of anticoagulation assessed by INR, suggesting that thrombin generation may be a useful tool for monitoring ongoing hypercoagulable states in patients with APS on anticoagulation [147••]. Although thrombin generation assays are largely limited to the research setting, they may prove useful for monitoring anticoagulation efficacy in thrombotic APS, particularly for those who develop recurrent thromboses despite anticoagulation.
Activation of vascular cells by aPL effects is central to the pathogenesis of APS. Microparticles, submicron particles released from all cells in response to stimuli such as cellular activation and/or apoptosis [148], have been evaluated as a biomarker in APS. Several studies have reported elevated numbers of circulating endothelial cell- and platelet-derived microparticles in patients with APS [149,150,151,152,153,154,155]. MP from APS plasma also demonstrate elevated TF activity [156]. While some studies noted a correlation between levels of MP and thrombotic complications [150, 153], others have failed to demonstrate this association [149, 154, 155]. In patients with aPL, elevated levels of MP are present remote from the time of thrombotic events, and anticoagulant therapy does not reduce MP levels, indicating that anticoagulation masks but does not address the chronic pro-inflammatory and prothrombotic state underlying APS. Microparticles are an attractive candidate biomarker in APS, both to predict thrombotic risk and to monitor efficacy of therapy. However, MP measurements have been plagued by a lack of standardized methodology for isolation, quantification, and functional analyses, as well as lack of reproducibility of measurements in individual patients on repeated testing. Concerted efforts to address these issues are needed.
Given the role of complement in the pathogenesis of APS-related complications, complement markers have been evaluated as biomarkers in APS. In primary APS, hypocomplementemia may be associated with LA, as well as livedo reticularis and thrombocytopenia [157]. While increased levels of complement activation products, indicating complement activation, have been reported in patients with primary APS, data regarding their correlation with thrombosis are conflicting [158, 159]. Complement activation (elevated alternate pathway convertase C3bBbP terminal complement components sC5b-9) has been demonstrated and is likely involved in the pathogenesis of catastrophic APS [157]. Developing validated assays for complement activation through measuring complement activation products or using novel functional assays [160] could potentially aid in the diagnosis of catastrophic APS, and also might predict responses to eculizumab in this disorder. Others have evaluated gene expression signatures and proteomic approaches to predicting the risk of clinical events [161]. These novel approaches may yield clinically useful markers and insights into pathophysiology.
A relatively unexplored area of research is the role of genetic predisposition in APS-mediated clinical events. It is possible that polymorphisms in key proteins involved in anti-β2GPI antibody-mediated signaling or effector pathways may render certain individuals more susceptible to thrombosis or pregnancy loss induced by these antibodies. For example, one study demonstrated that mice in which the inflammatory response to LPS was absent due to a missense point mutation in the cytoplasmic tail of TLR4 did not display enhanced thrombosis after passive infusion of human aPL [33], while those with wild-type TLR4 did. Moreover, co-segregating TLR4 Asp299Gly and Thr399Ile polymorphisms were found to occur with lower frequency in patients with APS vs. healthy controls; however, the frequency in patients with aPL without thrombosis was not determined.
Conclusions
Tools to individualize thrombotic risk assessment are critical for the optimal management of individuals with persistent aPL, with or without APS-related complications. Current approaches are mostly limited to the aPL profile and traditional thrombotic risk factors. Recent developments include domain 1 specific anti-β2GPI antibodies, other aPL, clinical risk calculators, and “biologic” assays based on pathophysiology such as thrombin generation and complement activation. Prospective studies will be needed to design and validate layered approaches that integrate standard diagnostic criteria with newer analytic assays to improve APS diagnosis. In addition, laboratory strategies to identify patients that can safely be treated with a shorter duration of anticoagulation, and those with persistent hypercoagulable states on anticoagulation, would be useful. Concerted efforts are required to validate, standardize, and implement these promising new strategies for patients with APS.
References
Recently published papers of particular interest have been highlighted as: • Of importance, •• Of major importance
• Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306. This paper describes the current clinical and laboratory diagnostic criteria for APS
Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46(4):1019–27.
Cervera R, Espinosa G. Update on the catastrophic antiphospholipid syndrome and the “CAPS Registry”. Semin Thromb Hemost. 2012;38(4):333–8.
Asherson RA, Cervera R, de Groot PG, Erkan D, Boffa MC, Piette JC, et al. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12(7):530–4.
Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramon E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2015;74(6):1011–8.
Brey RL, Muscal E, Chapman J. Antiphospholipid antibodies and the brain: a consensus report. Lupus. 2011;20(2):153–7.
Urbanus RT, de Laat B. Antiphospholipid antibodies and the protein C pathway. Lupus. 2010;19(4):394–9.
Rossetto V, Spiezia L, Franz F, Salmaso L, Pozza LV, Gavasso S, et al. The role of antiphospholipid antibodies toward the protein C/protein S system in venous thromboembolic disease. Am J Hematol. 2009;84(9):594–6.
Wahl D, Membre A, Perret-Guillaume C, Regnault V, Lecompte T. Mechanisms of antiphospholipid-induced thrombosis: effects on the protein C system. Curr Rheumatol Rep. 2009;11(1):77–81.
Esmon CT. The anticoagulant and anti-inflammatory roles of the protein C anticoagulant pathway. J Autoimmun. 2000;15(2):113–6.
Marciniak E, Romond EH. Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood. 1989;74(7):2426–32.
Borrell M, Sala N, de Castellarnau C, Lopez S, Gari M, Fontcuberta J. Immunoglobulin fractions isolated from patients with antiphospholipid antibodies prevent the inactivation of factor Va by activated protein C on human endothelial cells. Thromb Haemost. 1992;68(3):268–72.
de Laat B, Eckmann CM, van Schagen M, Meijer AB, Mertens K, van Mourik JA. Correlation between the potency of a beta2-glycoprotein I-dependent lupus anticoagulant and the level of resistance to activated protein C. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 2008;19(8):757–64.
Galli M, Willems GM, Rosing J, Janssen RM, Govers-Riemslag JW, Comfurius P, et al. Anti-prothrombin IgG from patients with anti-phospholipid antibodies inhibits the inactivation of factor Va by activated protein C. Br J Haematol. 2005;129(2):240–7.
Nojima J, Kuratsune H, Suehisa E, Iwatani Y, Kanakura Y. Acquired activated protein C resistance associated with IgG antibodies against beta2-glycoprotein I and prothrombin as a strong risk factor for venous thromboembolism. Clin Chem. 2005;51(3):545–52.
Izumi T, Pound ML, Su Z, Iverson GM, Ortel TL. Anti-beta(2)-glycoprotein I antibody-mediated inhibition of activated protein C requires binding of beta(2)-glycoprotein I to phospholipids. Thromb Haemost. 2002;88(4):620–6.
Smirnov MD, Triplett DT, Comp PC, Esmon NL, Esmon CT. On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest. 1995;95(1):309–16.
Shibata S, Harpel PC, Gharavi A, Rand J, Fillit H. Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood. 1994;83(9):2532–40.
Liestol S, Sandset PM, Jacobsen EM, Mowinckel MC, Wisloff F. Decreased anticoagulant response to tissue factor pathway inhibitor type 1 in plasmas from patients with lupus anticoagulants. Br J Haematol. 2007;136(1):131–7.
Bu C, Gao L, Xie W, Zhang J, He Y, Cai G, et al. Beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation. Arthritis Rheum. 2009;60(2):559–68.
Rand JH, Wu XX, Quinn AS, Chen PP, McCrae KR, Bovill EG, et al. Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay. Am J Pathol. 2003;163(3):1193–200.
Rand JH, Wu XX, Quinn AS, Chen PP, Hathcock JJ, Taatjes DJ. Hydroxychloroquine directly reduces the binding of antiphospholipid antibody-beta2-glycoprotein I complexes to phospholipid bilayers. Blood. 2008;112(5):1687–95.
Giannakopoulos B, Passam F, Rahgozar S, Krilis SA. Current concepts on the pathogenesis of the antiphospholipid syndrome. Blood. 2007;109(2):422–30.
Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–44.
Simantov R, LaSala JM, Lo SK, Gharavi AE, Sammaritano LR, Salmon JE, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96(5):2211–9.
Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood. 2005;105(5):1964–9.
Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, Ichikawa K, et al. Endothelial cells as target for antiphospholipid antibodies. Human polyclonal and monoclonal anti-beta 2-glycoprotein I antibodies react in vitro with endothelial cells through adherent beta 2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 1997;40(3):551–61.
Branch DW, Rodgers GM. Induction of endothelial cell tissue factor activity by sera from patients with antiphospholipid syndrome: a possible mechanism of thrombosis. Am J Obstet Gynecol. 1993;168(1 Pt 1):206–10.
Mineo C. Inhibition of nitric oxide and antiphospholipid antibody-mediated thrombosis. Curr Rheumatol Rep. 2013;15(5):324.
Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood. 2012;119(3):884–93.
Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J Clin Invest. 2011;121(1):120–31.
• Wu M, Barnard J, Kundu S, McCrae KR. A novel pathway of cellular activation mediated by antiphospholipid antibody-induced extracellular vesicles. J Thromb Haemost. 2015;13(10):1928–40. This study demonstrated a novel pathway of cellular activation mediated by aPL-induced extracellular vesicles and IL-1 receptor-associated kinase 4 phosphorylation.
Pierangeli SS, Vega-Ostertag ME, Raschi E, Liu X, Romay-Penabad Z, De Micheli V, et al. Toll-like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis. 2007;66(10):1327–33.
Romay-Penabad Z, Montiel-Manzano MG, Shilagard T, Papalardo E, Vargas G, Deora AB, et al. Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood. 2009;114(14):3074–83.
Romay-Penabad Z, Aguilar-Valenzuela R, Urbanus RT, Derksen RH, Pennings MT, Papalardo E, et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. 2011;117(4):1408–14.
Montiel-Manzano G, Romay-Penabad Z, PapalardodeMartinez E, Meillon-Garcia LA, Garcia-Latorre E, Reyes-Maldonado E et al. In vivo effects of an inhibitor of nuclear factor-kappa B on thrombogenic properties of antiphospholipid antibodies. Ann N Y Acad Sci 2007;1108:540–553.
Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. 2007;56(8):2687–97.
Vega-Ostertag M, Harris EN, Pierangeli SS. Intracellular events in platelet activation induced by antiphospholipid antibodies in the presence of low doses of thrombin. Arthritis Rheum. 2004;50(9):2911–9.
Pennings MT, Derksen RH, van Lummel M, Adelmeijer J, VanHoorelbeke K, Urbanus RT, et al. Platelet adhesion to dimeric beta-glycoprotein I under conditions of flow is mediated by at least two receptors: glycoprotein Ibalpha and apolipoprotein E receptor 2′. Journal of thrombosis and haemostasis : JTH. 2007;5(2):369–77.
Shi T, Giannakopoulos B, Yan X, Yu P, Berndt MC, Andrews RK, et al. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. 2006;54(8):2558–67.
Mulla MJ, Brosens JJ, Chamley LW, Giles I, Pericleous C, Rahman A, et al. Antiphospholipid antibodies induce a pro-inflammatory response in first trimester trophoblast via the TLR4/MyD88 pathway. American journal of reproductive immunology (New York, NY : 1989). 2009;62(2):96–111.
Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112(11):1644–54.
Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. 2004;10(11):1222–6.
Salmon JE, Girardi G, Holers VM. Activation of complement mediates antiphospholipid antibody-induced pregnancy loss. Lupus. 2003;12(7):535–8.
Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. 2005;106(7):2340–6.
Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum. 2005;52(7):2120–4.
•• Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to beta2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. 2014;123(22):3478–87. In this study, the authors demonstrated a dominant role of complement activation in APS by showing that a CH2 deleted aPL prevented and reversed aPL-induced thrombosis and pregnancy loss in a murine model.
Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794–802.
Redecha P, Tilley R, Tencati M, Salmon JE, Kirchhofer D, Mackman N, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110(7):2423–31.
• Barratt-Due A, Floisand Y, Orrem HL, Kvam AK, Holme PA, Bergseth G, et al. Complement activation is a crucial pathogenic factor in catastrophic antiphospholipid syndrome. Rheumatology (Oxford). 2016;55(7):1337–9. This study demonstrates complement activation, measured by increased C4bc/C4 ratio (classical pathway), C3bBbP (alternate pathway), and C5b-9 complex (terminal pathway) in a patient with APS.
• Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. J Clin Rheumatol. 2015;21(6):311–3. This report describes the successful treatment of refractory CAPS with eculizumab, with no recurrence over 16 months of follow up.
Shapira I, Andrade D, Allen SL, Salmon JE. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64(8):2719–23.
•• Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14(2):459–65. This study demonstrates the successful use of eculizumab, a humanized monoclonal antibody against terminal complement component C5, in patients with CAPS and renal failure requiring transplantation.
Wig S, Chan M, Thachil J, Bruce I, Barnes T. A case of relapsing and refractory catastrophic anti-phospholipid syndrome successfully managed with eculizumab, a complement 5 inhibitor. Rheumatology (Oxford). 2016 Feb;55(2):382–4.
Kronbichler A, Frank R, Kirschfink M, Szilagyi A, Csuka D, Prohaszka Z, et al. Efficacy of eculizumab in a patient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: a case report. Medicine (Baltimore). 2014;93(26):e143.
Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42(7):1309–11.
Pengo V, Tripodi A, Reber G, Rand JH, Ortel TL, Galli M, et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7(10):1737–40.
Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost. 1995;74(4):1185–90.
Ruiz-Garcia R, Serrano M, Martinez-Flores JA, Mora S, Morillas L, Martin-Mola MA, et al. Isolated IgA anti- beta2 glycoprotein I antibodies in patients with clinical criteria for antiphospholipid syndrome. J Immunol Res. 2014;2014:704395.
• Cousins L, Pericleous C, Khamashta M, Bertolaccini ML, Ioannou Y, Giles I, et al. Antibodies to domain I of beta-2-glycoprotein I and IgA antiphospholipid antibodies in patients with ‘seronegative’ antiphospholipid syndrome. Ann Rheum Dis. 2015;74(1):317–9. This study describes the presence of domain 1 anti-β 2 GPI and IgA aPL in patients with clinical signs of APS but negative for standard serologic assays, often termed “seronegative APS.”
Mattia E, Ruffatti A, Tonello M, Meneghel L, Robecchi B, Pittoni M, et al. IgA anticardiolipin and IgA anti-beta2 glycoprotein I antibody positivity determined by fluorescence enzyme immunoassay in primary antiphospholipid syndrome. Clin Chem Lab Med. 2014;52(9):1329–33.
Nayfe R, Uthman I, Aoun J, Saad Aldin E, Merashli M, Khamashta MA. Seronegative antiphospholipid syndrome. Rheumatology (Oxford). 2013;52(8):1358–67.
• Conti F, Capozzi A, Truglia S, Lococo E, Longo A, Misasi R, et al. The mosaic of “seronegative” antiphospholipid syndrome. J Immunol Res. 2014;2014:389601. This study describes a number of aPL against alternative phospholpid antigens such as annexin V, propthrombin, vimentin/cardiolipin complexes, etc. in patients with seronegative APS.
Salle V, Maziere JC, Smail A, Cevallos R, Maziere C, Fuentes V, et al. Anti-annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J Clin Immunol. 2008;28(4):291–7.
Cesarman-Maus G, Rios-Luna NP, Deora AB, Huang B, Villa R, Cravioto Mdel C, et al. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood. 2006;107(11):4375–82.
Becarevic M. The IgG and IgM isotypes of anti-annexin A5 antibodies: relevance for primary antiphospholipid syndrome. J Thromb Thrombolysis. 2016;42(4):552–7.
Ortona E, Capozzi A, Colasanti T, Conti F, Alessandri C, Longo A, et al. Vimentin/cardiolipin complex as a new antigenic target of the antiphospholipid syndrome. Blood. 2010;116(16):2960–7.
Petri M. Epidemiology of the antiphospholipid antibody syndrome. J Autoimmun. 2000;15(2):145–51.
Galli M, Luciani D, Bertolini G, Barbui T. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome: a systematic review of the literature. Blood. 2003;101(5):1827–32.
Galli M, Borrelli G, Jacobsen EM, Marfisi RM, Finazzi G, Marchioli R, et al. Clinical significance of different antiphospholipid antibodies in the WAPS (warfarin in the antiphospholipid syndrome) study. Blood. 2007;110(4):1178–83.
Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol. 2009;8(11):998–1005.
Galli M, Luciani D, Bertolini G, Barbui T. Anti-beta 2-glycoprotein I, antiprothrombin antibodies, and the risk of thrombosis in the antiphospholipid syndrome. Blood. 2003;102(8):2717–23.
de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost. 2005;3(9):1993–7.
Urbanus RT, de Groot PG. Antiphospholipid antibodies—we are not quite there yet. Blood Rev. 2011;25(2):97–106.
Devreese K, Hoylaerts MF. Challenges in the diagnosis of the antiphospholipid syndrome. Clin Chem. 2010;56(6):930–40.
Opatrny L, David M, Kahn SR, Shrier I, Rey E. Association between antiphospholipid antibodies and recurrent fetal loss in women without autoimmune disease: a metaanalysis. J Rheumatol. 2006;33(11):2214–21.
de Laat HB, Derksen RH, Urbanus RT, Roest M, de Groot PG. Beta2-glycoprotein I-dependent lupus anticoagulant highly correlates with thrombosis in the antiphospholipid syndrome. Blood. 2004;104(12):3598–602.
Pengo V, Ruffatti A, Legnani C, Gresele P, Barcellona D, Erba N, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost. 2010;8(2):237–42.
Pengo V, Ruffatti A, Legnani C, Testa S, Fierro T, Marongiu F, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood. 2011;118(17):4714–8.
Lee EY, Lee CK, Lee TH, Chung SM, Kim SH, Cho YS, et al. Does the anti-beta2-glycoprotein I antibody provide additional information in patients with thrombosis? Thromb Res. 2003;111(1–2):29–32.
• Yelnik CM, Urbanski G, Drumez E, Sobanski V, Maillard H, Lanteri A, et al. Persistent triple antiphospholipid antibody positivity as a strong risk factor of first thrombosis, in a long-term follow-up study of patients without history of thrombosis or obstetrical morbidity. Lupus. 2016 Jul 17. This study confirmed that asymptomatic aPL carriers, especially those that are “triple positive,” have a higher rate of first thrombotic or obstetric events but failed to demonstrate a benefit of aspirin prophylaxis.
• Mustonen P, Lehtonen KV, Javela K, Puurunen M. Persistent antiphospholipid antibody (aPL) in asymptomatic carriers as a risk factor for future thrombotic events: a nationwide prospective study. Lupus. 2014;23(14):1468–76. This study also confirmed that asymptomatic aPL carriers have a high risk of future APS-related clinical events, which is increased in the presence of SLE or other autoimmune disease.
Ruffatti A, Tonello M, Del Ross T, Cavazzana A, Grava C, Noventa F, et al. Antibody profile and clinical course in primary antiphospholipid syndrome with pregnancy morbidity. Thromb Haemost. 2006;96(3):337–41.
Pierangeli SS, Liu XW, Barker JH, Anderson G, Harris EN. Induction of thrombosis in a mouse model by IgG, IgM and IgA immunoglobulins from patients with the antiphospholipid syndrome. Thromb Haemost. 1995;74(5):1361–7.
Diri E, Cucurull E, Gharavi AE, Kapoor D, Mendez EA, Scopelitis E, et al. Antiphospholipid (Hughes’) syndrome in African-Americans: IgA aCL and abeta2 glycoprotein-I is the most frequent isotype. Lupus. 1999;8(4):263–8.
Danowski A, Kickler TS, Petri M. Anti-beta2-glycoprotein I: prevalence, clinical correlations, and importance of persistent positivity in patients with antiphospholipid syndrome and systemic lupus erythematosus. J Rheumatol. 2006;33(9):1775–9.
Mehrani T, Petri M. Association of IgA anti-beta2 glycoprotein I with clinical and laboratory manifestations of systemic lupus erythematosus. J Rheumatol. 2011;38(1):64–8.
Sweiss NJ, Bo R, Kapadia R, Manst D, Mahmood F, Adhikari T, et al. IgA anti-beta2-glycoprotein I autoantibodies are associated with an increased risk of thromboembolic events in patients with systemic lupus erythematosus. PLoS One. 2010;5(8):e12280.
Meroni PL, Chighizola CB, Rovelli F, Gerosa M. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16(2):209.
Hoylaerts MF. To the heart of the APS puzzle. Blood. 2010;116(16):2867–9.
de Laat B, Derksen RH, Urbanus RT, de Groot PG. IgG antibodies that recognize epitope Gly40-Arg43 in domain I of beta 2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood. 2005;105(4):1540–5.
de Laat B, Pengo V, Pabinger I, Musial J, Voskuyl AE, Bultink IE, et al. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009 Nov;7(11):1767–73.
•• Pengo V, Ruffatti A, Tonello M, Cuffaro S, Banzato A, Bison E, et al. Antiphospholipid syndrome: antibodies to Domain 1 of beta2-glycoprotein 1 correctly classify patients at risk. J Thromb Haemost. 2015;13(5):782–7. This study showed that IgG anti-β2GPI-Domain1 titers are significantly higher in individuals with a triple-positive aPL profile, and that there is an association between the concentration of these antibodies and thrombosis risk categories.
De Craemer AS, Musial J, Devreese KM. Role of anti-domain 1-beta2 glycoprotein I antibodies in the diagnosis and risk stratification of antiphospholipid syndrome. J Thromb Haemost. 2016;14(9):1779–87.
Mahler M, Norman GL, Meroni PL, Khamashta M. Autoantibodies to domain 1 of beta 2 glycoprotein 1: a promising candidate biomarker for risk management in antiphospholipid syndrome. Autoimmun Rev. 2012;12(2):313–7.
Galli M, Barbui T. Prothrombin as cofactor for antiphospholipids. Lupus. 1998;7(Suppl 2):S37–40.
Fleck RA, Rapaport SI, Rao LV. Anti-prothrombin antibodies and the lupus anticoagulant. Blood. 1988;72(2):512–9.
Erkan D, Bateman H, Lockshin MD. Lupus anticoagulant-hypoprothrombinemia syndrome associated with systemic lupus erythematosus: report of 2 cases and review of literature. Lupus. 1999;8(7):560–4.
Simmelink MJ, Horbach DA, Derksen RH, Meijers JC, Bevers EM, Willems GM, et al. Complexes of anti-prothrombin antibodies and prothrombin cause lupus anticoagulant activity by competing with the binding of clotting factors for catalytic phospholipid surfaces. Br J Haematol. 2001;113(3):621–9.
Horbach DA, van Oort E, Derksen RH, de Groot PG. The contribution of anti-prothrombin-antibodies to lupus anticoagulant activity—discrimination between functional and non-functional anti-prothrombin-antibodies. Thromb Haemost. 1998;79(4):790–5.
Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal RF. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. 1991;66(6):629–32.
Sciascia S, Khamashta MA, Bertolaccini ML. New tests to detect antiphospholipid antibodies: antiprothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies. Curr Rheumatol Rep. 2014;16(5):415.
Bertolaccini ML, Atsumi T, Khamashta MA, Amengual O, Hughes GR. Autoantibodies to human prothrombin and clinical manifestations in 207 patients with systemic lupus erythematosus. J Rheumatol. 1998;25(6):1104–8.
Bertolaccini ML, Atsumi T, Koike T, Hughes GR, Khamashta MA. Antiprothrombin antibodies detected in two different assay systems. Prevalence and clinical significance in systemic lupus erythematosus. Thromb Haemost. 2005;93(2):289–97.
Tsutsumi A, Hayashi T, Chino Y, Mamura M, Goto D, Matsumoto I, et al. Significance of antiprothrombin antibodies in patients with systemic lupus erythematosus: clinical evaluation of the antiprothrombin assay and the antiphosphatidylserine/prothrombin assay, and comparison with other antiphospholipid antibody assays. Mod Rheumatol. 2006;16(3):158–64.
Pengo V, Denas G, Bison E, Banzato A, Jose SP, Gresele P, et al. Prevalence and significance of anti-prothrombin (aPT) antibodies in patients with lupus anticoagulant (LA). Thromb Res. 2010;126(2):150–3.
Galli M, Beretta G, Daldossi M, Bevers EM, Barbui T. Different anticoagulant and immunological properties of anti-prothrombin antibodies in patients with antiphospholipid antibodies. Thromb Haemost. 1997;77(3):486–91.
Atsumi T, Koike T. Antiprothrombin antibody: why do we need more assays? Lupus. 2010;19(4):436–9.
Zigon P, Ambrozic A, Cucnik S, Kveder T, Rozman B, Bozic B. Modified phosphatidylserine-dependent antiprothrombin [corrected] ELISA enables identification of patients negative for other antiphospholipid antibodies and also detects low avidity antibodies. Clin Chem Lab Med. 2011;49(6):1011–8.
Vlagea A, Gil A, Cuesta MV, Arribas F, Diez J, Lavilla P, et al. Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential markers of antiphospholipid syndrome. Clin Appl Thromb Hemost. 2013;19(3):289–96.
Pregnolato F, Chighizola CB, Encabo S, Shums Z, Norman GL, Tripodi A, et al. Anti-phosphatidylserine/prothrombin antibodies: an additional diagnostic marker for APS? Immunol Res. 2013;56(2–3):432–8.
Sciascia S, Sanna G, Murru V, Roccatello D, Khamashta MA, Bertolaccini ML. Anti-prothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome. A systematic review. Thromb Haemost. 2014;111(2):354–64.
Sciascia S, Murru V, Sanna G, Roccatello D, Khamashta MA, Bertolaccini ML. Clinical accuracy for diagnosis of antiphospholipid syndrome in systemic lupus erythematosus: evaluation of 23 possible combinations of antiphospholipid antibody specificities. J Thromb Haemost. 2012;10(12):2512–8.
Heikal NM, Jaskowski TD, Malmberg E, Lakos G, Branch DW, Tebo AE. Laboratory evaluation of anti-phospholipid syndrome: a preliminary prospective study of phosphatidylserine/prothrombin antibodies in an at-risk patient cohort. Clin Exp Immunol. 2015;180(2):218–26.
Inanc M, Donohoe S, Ravirajan CT, Radway-Bright EL, Mackie I, Machin S, et al. Anti-beta2-glycoprotein I, anti-prothrombin and anticardiolipin antibodies in a longitudinal study of patients with systemic lupus erythematosus and the antiphospholipid syndrome. Br J Rheumatol. 1998;37(10):1089–94.
•• Reynaud Q, Lega JC, Mismetti P, Chapelle C, Wahl D, Cathebras P, et al. Risk of venous and arterial thrombosis according to type of antiphospholipid antibodies in adults without systemic lupus erythematosus: a systematic review and meta-analysis. Autoimmun Rev. 2014;13(6):595–608. This large metanalysis and systematic review including 30 studies with 16.441 patients demonstrated that LA were stronger risk factors for venous thrombosis than aCL. For arterial thrombosis, LA were the strongest risk factors, followed by anti-β2GPI, aPS, aPT, and then aCL.
Staub HL, Bertolaccini ML, Khamashta MA. Anti-phosphatidylethanolamine antibody, thromboembolic events and the antiphospholipid syndrome. Autoimmun Rev. 2012;12(2):230–4.
Karmochkine M, Cacoub P, Piette JC, Godeau P, Boffa MC. Antiphosphatidylethanolamine antibody as the sole antiphospholipid antibody in systemic lupus erythematosus with thrombosis. Clin Exp Rheumatol. 1992;10(6):603–5.
Balada E, Ordi-Ros J, Paredes F, Villarreal J, Mauri M, Vilardell-Tarres M. Antiphosphatidylethanolamine antibodies contribute to the diagnosis of antiphospholipid syndrome in patients with systemic lupus erythematosus. Scand J Rheumatol. 2001;30(4):235–41.
Karmochkine M, Berard M, Piette JC, Cacoub P, Aillaud MF, Harle JR, et al. Antiphosphatidylethanolamine antibodies in systemic lupus erythematosus. Lupus. 1993;2(3):157–60.
Hirmerova J, Ulcova-Gallova Z, Seidlerova J, Filipovsky J, Bibkova K, Micanova Z, et al. Laboratory evaluation of antiphospholipid antibodies in patients with venous thromboembolism. Clin Appl Thromb Hemost. 2010;16(3):318–25.
Sanmarco M, Gayet S, Alessi MC, Audrain M, de Maistre E, Gris JC, et al. Antiphosphatidylethanolamine antibodies are associated with an increased odds ratio for thrombosis. A multicenter study with the participation of the European Forum on antiphospholipid antibodies. Thromb Haemost. 2007;97(6):949–54.
Berard M, Boffa MC, Karmochkine M, Aillaud MF, Juhan-Vague I, Frances C, et al. Plasma reactivity to hexagonal II phase phosphatidylethanolamine is more frequently associated with lupus anticoagulant than with antiphosphatidylethanolamine antibodies. J Lab Clin Med. 1993;122(5):601–5.
Rauch J, Tannenbaum M, Neville C, Fortin PR. Inhibition of lupus anticoagulant activity by hexagonal phase phosphatidylethanolamine in the presence of prothrombin. Thromb Haemost. 1998;80(6):936–41.
Rand JH, Wu XX, Andree HA, Ross JB, Rusinova E, Gascon-Lema MG, et al. Antiphospholipid antibodies accelerate plasma coagulation by inhibiting annexin-V binding to phospholipids: a “lupus procoagulant” phenomenon. Blood. 1998;92(5):1652–60.
Rand JH, Wu XX, Lapinski R, van Heerde WL, Reutelingsperger CP, Chen PP, et al. Detection of antibody-mediated reduction of annexin A5 anticoagulant activity in plasmas of patients with the antiphospholipid syndrome. Blood. 2004;104(9):2783–90.
de Laat B, Derksen RH, Mackie IJ, Roest M, Schoormans S, Woodhams BJ, et al. Annexin A5 polymorphism (−1C-->T) and the presence of anti-annexin A5 antibodies in the antiphospholipid syndrome. Ann Rheum Dis. 2006;65(11):1468–72.
Nojima J, Kuratsune H, Suehisa E, Futsukaichi Y, Yamanishi H, Machii T, et al. Association between the prevalence of antibodies to beta(2)-glycoprotein I, prothrombin, protein C, protein S, and annexin V in patients with systemic lupus erythematosus and thrombotic and thrombocytopenic complications. Clin Chem. 2001;47(6):1008–15.
Arnold J, Holmes Z, Pickering W, Farmer C, Regan L, Cohen H. Anti-beta 2 glycoprotein 1 and anti-annexin V antibodies in women with recurrent miscarriage. Br J Haematol. 2001;113(4):911–4.
Blaschek MA, Boehme M, Jouquan J, Simitzis AM, Fifas S, Le Goff P, et al. Relation of antivimentin antibodies to anticardiolipin antibodies in systemic lupus erythematosus. Ann Rheum Dis. 1988;47(9):708–16.
Otomo K, Atsumi T, Amengual O, Fujieda Y, Kato M, Oku K, et al. Efficacy of the antiphospholipid score for the diagnosis of antiphospholipid syndrome and its predictive value for thrombotic events. Arthritis Rheum. 2012;64(2):504–12.
Sciascia S, Bertolaccini ML, Roccatello D, Khamashta MA. Independent validation of the antiphospholipid score for the diagnosis of antiphospholipid syndrome. Ann Rheum Dis. 2013;72(1):142–3.
Sciascia S, Sanna G, Murru V, Roccatello D, Khamashta MA, Bertolaccini ML. GAPSS: the global anti-phospholipid syndrome score. Rheumatology (Oxford, England). 2013;52(8):1397–403.
• Sciascia S, Sanna G, Murru V, Roccatello D, Khamashta MA, Bertolaccini ML. The global anti-phospholipid syndrome score in primary APS. Rheumatology (Oxford, England). 2015;54(1):134–138. This investigation validated the GAPSS tool in primary APS.
Oku K, Amengual O, Bohgaki T, Horita T, Yasuda S, Atsumi T. An independent validation of the global anti-phospholipid syndrome score in a Japanese cohort of patients with autoimmune diseases. Lupus. 2015;24(7):774–5.
Ruffatti A, Tonello M, Visentin MS, Bontadi A, Hoxha A, De Carolis S, et al. Risk factors for pregnancy failure in patients with anti-phospholipid syndrome treated with conventional therapies: a multicentre, case-control study. Rheumatology (Oxford). 2011;50(9):1684–9.
Buyon JP, Kim MY, Guerra MM, Laskin CA, Petri M, Lockshin MD, et al. Predictors of pregnancy outcomes in patients with lupus: a cohort study. Ann Intern Med. 2015;163(3):153–63.
Caruso A, De Carolis S, Ferrazzani S, Valesini G, Caforio L, Mancuso S. Pregnancy outcome in relation to uterine artery flow velocity waveforms and clinical characteristics in women with antiphospholipid syndrome. Obstet Gynecol. 1993;82(6):970–7.
Venkat-Raman N, Backos M, Teoh TG, Lo WT, Regan L. Uterine artery Doppler in predicting pregnancy outcome in women with antiphospholipid syndrome. Obstet Gynecol. 2001;98(2):235–42.
Hemker HC, Al Dieri R, De Smedt E, Beguin S. Thrombin generation, a function test of the haemostatic-thrombotic system. Thromb Haemost. 2006;96(5):553–61.
van Veen JJ, Gatt A, Makris M. Thrombin generation testing in routine clinical practice: are we there yet? Br J Haematol. 2008;142(6):889–903.
Dienava-Verdoold I, Boon-Spijker MG, de Groot PG, Brinkman HJ, Voorberg J, Mertens K, et al. Patient-derived monoclonal antibodies directed towards beta2 glycoprotein-1 display lupus anticoagulant activity. J Thromb Haemost. 2011;9(4):738–47.
Regnault V, Beguin S, Wahl D, de Maistre E, Coenraad Hemker H, Lecompte T. Thrombinography shows acquired resistance to activated protein C in patients with lupus anticoagulants. Thromb Haemost. 2003;89(2):208–12.
Devreese K, Peerlinck K, Arnout J, Hoylaerts MF. Laboratory detection of the antiphospholipid syndrome via calibrated automated thrombography. Thromb Haemost. 2009;101(1):185–96.
Hanly JG, Smith SA. Anti-beta2-glycoprotein I autoantibodies, in vitro thrombin generation, and the antiphospholipid syndrome. J Rheumatol. 2000;27(9):2152–9.
Devreese K, Peerlinck K, Hoylaerts MF. Thrombotic risk assessment in the antiphospholipid syndrome requires more than the quantification of lupus anticoagulants. Blood. 2010;115(4):870–8.
•• Efthymiou M, Lawrie AS, Mackie I, Arachchillage D, Lane PJ, Machin S, et al. Thrombin generation and factor X assays for the assessment of warfarin anticoagulation in thrombotic antiphospholipid syndrome. Thromb Res. 2015;135(6):1191–7. In this study, the authors demonstrated that some patients with APS on therapeutic anticoagulation with warfarin had persistent hypercoagulability measured in terms of increased endogenous thrombin potential and peak thrombin generation on thrombin generation assays suggesting that thrombin generation could be a useful tool for measuring hypercoagulability in anticoagulated patients with APS.
Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cellular and molecular life sciences : CMLS. 2011;68(16):2667–88.
Vikerfors A, Mobarrez F, Bremme K, Holmstrom M, Agren A, Eelde A, et al. Studies of microparticles in patients with the antiphospholipid syndrome (APS). Lupus. 2012;21(7):802–5.
Jy W, Tiede M, Bidot CJ, Horstman LL, Jimenez JJ, Chirinos J, et al. Platelet activation rather than endothelial injury identifies risk of thrombosis in subjects positive for antiphospholipid antibodies. Thromb Res. 2007;121(3):319–25.
Antwi-Baffour S, Kholia S, Aryee YK, Ansa-Addo EA, Stratton D, Lange S, et al. Human plasma membrane-derived vesicles inhibit the phagocytosis of apoptotic cells—possible role in SLE. Biochem Biophys Res Commun. 2010;398(2):278–83.
Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol. 2001;115(2):451–9.
Combes V, Simon AC, Grau GE, Arnoux D, Camoin L, Sabatier F, et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest. 1999;104(1):93–102.
Dignat-George F, Camoin-Jau L, Sabatier F, Arnoux D, Anfosso F, Bardin N, et al. Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost. 2004;91(4):667–73.
Chaturvedi S, Cockrell E, Espinola R, Hsi L, Fulton S, Khan M, et al. Circulating microparticles in patients with antiphospholipid antibodies: characterization and associations. Thromb Res. 2015;135(1):102–8.
Willemze R, Bradford RL, Mooberry MJ, Roubey RA, Key NS. Plasma microparticle tissue factor activity in patients with antiphospholipid antibodies with and without clinical complications. Thromb Res. 2014;133(2):187–9.
Ramos-Casals M, Campoamor MT, Chamorro A, Salvador G, Segura S, Botero JC, et al. Hypocomplementemia in systemic lupus erythematosus and primary antiphospholipid syndrome: prevalence and clinical significance in 667 patients. Lupus. 2004;13(10):777–83.
Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ. Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost. 2012;107(3):423–9.
Sarmiento E, Dale J, Arraya M, Gallego A, Lanio N, Navarro J, et al. CD8+DR+ T-cells and C3 complement serum concentration as potential biomarkers in thrombotic antiphospholipid syndrome. Autoimmune Dis. 2014;2014:868652.
Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol. 2013;56(3):232–9.
Lopez-Pedrera C, Cuadrado MJ, Herandez V, Buendia P, Aguirre MA, Barbarroja N, et al. Proteomic analysis in monocytes of antiphospholipid syndrome patients: deregulation of proteins related to the development of thrombosis. Arthritis Rheum. 2008;58(9):2835–44.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Shruti Chaturvedi and Keith R. McCrae declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Antiphospholipid Syndrome
Rights and permissions
About this article

Cite this article
Chaturvedi, S., McCrae, K.R. Clinical Risk Assessment in the Antiphospholipid Syndrome: Current Landscape and Emerging Biomarkers. Curr Rheumatol Rep 19, 43 (2017). https://doi.org/10.1007/s11926-017-0668-2
Published:
DOI: https://doi.org/10.1007/s11926-017-0668-2