
Abstract
Gout is the most common inflammatory arthropathy in the western world. Affecting millions and accounting for lost wages, increased health care costs, and significant disability, it remains a burden for those afflicted, their families, and the health care system. Despite the availability of a number of effective therapies, gout is often inadequately treated, and its impact on the patients overall health and well-being is underestimated by physicians and patients alike. For many decades, controlling acute flares was the priority in the management of gout. More recently, however, a deeper understanding of gout pathophysiology has resulted in a new appreciation that gout impacts the patient with consequences well beyond the episodes of acute inflammatory arthritis. Reflecting the chronic nature of the disease, gout treatment needs to be chronic as well, and aimed at reducing the underlying cause of gout—hyperuricemia—as well as the symptom of acute attacks. Therapy therefore requires both urate lowering and anti-inflammatory strategies. Unfortunately, the most commonly used urate lowering and anti-inflammatory treatments may be problematic in some gout patients, who often have multiple comorbidities that establish relative contraindications. Novel urate lowering therapies, and new medications to treat and prevent acute gouty flares, can not only improve care of the individual; they can also lead to a better discourse for the edification of those who manage and are managed for this underestimated disease. In this paper, we discuss new and pipeline drugs for acute gout, prophylactic anti-inflammatory therapies as well as urate lowering therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple drugs are currently in development for acute and chronic gout. The number of new agents being developed, and the specificity with which they act, reflect a robust commitment to exploring new scientific targets while expanding our knowledge and understanding of gout and associated comorbidities. Despite a plethora of advances in the knowledge and treatment of gout, a large number of patients continue to be inadequately managed. Although the majority of established gout treatments are inexpensive and generally effective, it is increasingly appreciated that for a significant minority of patients, these traditional therapies are inappropriate due to the presence of comorbidities, intolerance, unresponsiveness, or a combination of all three. Hypertension, cardiovascular disease, diabetes, renal impairment, peptic ulcer disease, metabolic syndrome, and liver disease are all common in gout patients and may create circumstances in which specific standard treatments are contraindicated. The inconvenience of long-term management via therapy titration, as is required for current urate lowering therapy, may also pose a compliance barrier for some patients and physicians.
The need for new therapies has led to renaissance in pharma gout research. GlobalData estimates that the acute gout treatment market will increase in size, reaching $337 million in 2018. The chronic gout market, which entails prophylactic anti-inflammatory therapies and urate lowering therapy, is expected to experience an even faster growth and will reach over $1.9 billion in 2018 [1]. Both the American College of Rheumatology and the European League Against Rheumatism have issued formal guidelines and recommendations for the treatment of gout. However, these guidelines do not encompass the needs of all categories of gout patients. To address the greatest unmet needs, pharmaceutical companies have been focusing their research on patients with more severe gout or with contraindications to current therapies, while attempting to minimize side effects and drug-drug interactions.
New Approaches to Gouty Inflammation
The Role of Interleukin-1β in Acute and Chronic Gout
Interleukin-1 (IL-1) is a proinflammatory cytokine that plays an essential role in mediating gouty inflammation [2••]. Monosodium urate (MSU) crystals directly activate the NLRP3 (NACHT, leucine-rich repeat and pyrin domain-containing protein-3) inflammasome through multiple possible mechanisms. MSU crystals released into the synovial fluid engage toll-like receptors on the surface of monocytes or macrophages, leading to cell activation. After MSU crystal phagocytosis, phagolysosomes are destabilized, which is associated with reactive oxygen species generation and decreased cytosolic potassium levels. These events cause activation of NLRP3 inflammasome, including the activation of pro-caspase-1 to caspase-1. Caspase-1 then cleaves pro-interleukin (IL)-1β to generate the mature active form. Active IL-1β is then secreted, resulting in the initial inflammatory response, which can further the activation of synovial lining cells and phagocytes [2••].
Based on our recent understanding of this biology, the use and/or development IL-1β inhibitors, and other compounds targeting the NLRP3 inflammasome, are a main developmental priority for addressing gouty inflammation (Fig. 1).

NLRP3 inflammasome and the main targets for current and pipeline therapies for gouty inflammation
Approved Biologics That Target Interleukin-1β
Three biologic agents are currently FDA approved to target IL-1: anakinra, rilonacept, and canakinumab. However, none of these agents are currently approved for gout, having instead received approval for Cryopyrin-Associated Periodic Syndromes (CAPS), systemic juvenile idiopathic arthritis (SJIA), and/or rheumatoid arthritis. Anakinra is an IL‑1 receptor antagonist (IL-1Ra) that engages the IL-1 receptor and blocks the binding of both IL 1α and IL‑1β, thus preventing signal transduction. Rilonacept is a soluble decoy receptor that binds and neutralizes IL-1β, preventing its interaction with cell surface IL-1 receptors. Finally, canakinumab is a monoclonal with IL-1β neutralizing capacity. Importantly, none of these agents have known adverse effects on hypertension, renal disease, or diabetes; all of which are common among gout patients.
Although no placebo-controlled or randomized controlled trials of anakinra in acute gout have been published to date, preliminary evidence and clinical reports support its efficacy in acute and subacute gout [3–5]. The optimal anakinra dosing, intervals, and length of treatment for both acute gout and subacute gout remain to be determined, but pilot studies suggest that a daily dose for 3–5 days may be sufficient to break gouty attacks.
To the best of our knowledge, rilonacept is no longer under study for gouty inflammation, having failed to show superiority to traditional NSAIDs. However, it should be noted that, in these studies, rilonacept was not inferior to the comparator NSAIDs.
Canakinumab has the longest half-life (23–26 days) of the three agents, which may be particularly valuable if a long-term effect is needed. Two phase III studies compared a single dose of canakinumab 150 mg subcutaneous (SC) with a single dose of intramuscular (IM) triamcinolone acetate (TA) for acute gouty attacks. Both studies were 12 weeks long in duration, followed by a 12-week extension. These studies found canakinumab 150 mg to provided more rapid pain relief as compared with TA 40 mg IM [6••].
In a randomized double-blind active-controlled multicenter trial assessing prophylaxis for gout attacks during initiation of urate lowering therapy (ULT), a single dose of canakinumab effectively reduced the risk of attacks in patients starting treatment with allopurinol [7]. All canakinumab doses investigated were superior to colchicine 0.5 mg once daily for attack prevention over the 16-week study period. At week 16, the mean number of gout attacks per patient was reduced by 62–72 % for canakinumab doses ≥50 mg compared with colchicine (p ≤ 0.0083). In addition, there was a 64–72 % reduction in the risk of experiencing ≥1 gout attack for canakinumab doses ≥50 mg compared with colchicine (p ≤ 0.05). The percentage of patients experiencing ≥1 gout attack was significantly lower for all canakinumab doses (15–27 %) compared with colchicine (44 %, p < 0.05). Taken together, these studies suggest that a single dose of canakinumab could potentially provide both acute treatment and durable prophylaxis in appropriate candidates.
In 2013, canakinumab became the first and is still the only IL-1 inhibitor approved in the European Union for treatment of patients with frequent gout attacks (≥3 attacks in the previous 12 months), in whom NSAIDs and colchicine are contraindicated, not tolerated, or do not provide an adequate response, and in whom repeated courses of corticosteroids (CS) are not appropriate [8]. The FDA Advisory Committee did not approve canakinumab use for gout concluding that treatment with canakinumab for acute gout is associated with an increased risk of infections, and that it is associated with decreased white blood cell counts, a mild decline in creatinine clearance and occurrence of hypertriglyceridemia, and that the potential for benefit therefore did not exceed the potential for risk [9].
Despite the lack of an FDA indication for these drugs, the 2012 American College of Rheumatology (ACR) recommendations for the management of gout propose the use of a biologic IL-1 inhibitor (anakinra 100 mg SC daily for 3 days or one injection of canakinumab 150 mg SC) as an option for severe gout refractory to standard treatment [10•].
Bucillamine: an Oral Anti-inflammatory Drug in Clinical Studies for Gout
Bucillamine (N-(2-mercapto-2-methylpropionyl)-L-cysteine) is a disease-modifying anti-rheumatic drug used as a first-line treatment for rheumatoid arthritis in Japan [11]. Bucillamine exerts an antioxidant effect by maintaining the endogenous glutaredoxin (Gtx) and thioredoxin (TRx) systems in a reduced state by transfer of thiol groups [12, 13]. In addition to its antioxidant action, bucillamine also increases the transcriptional activity of Nrf 2 [14].
In pre-clinical studies, bucillamine inhibited the release of IL-Ι β and IL-6 from mouse macrophages in response to MSU crystals and attenuated the release of TNF-α, IL-Ιβ, and IL-8 from THP-1 cells stimulated with lipopolysaccharides (LPS) [15]. These observations suggest that bucillamine might inhibit MSU crystal-induced inflammasome activation. It has been shown that uric acid triggers the association of NLRP3 with thioredoxin-interacting protein (TxNIP) in a reactive oxygen radical-dependent manner [16]. In unstimulated cells, TxNIP is constitutively bound to and inhibited by Trx. Following an increase in oxidative stress, this complex dissociates and TxNIP binds to NLRP3 promoting the assembly and oligomerization of the inflammasome. In support of such an activation mechanism, the knockdown of thioredoxin potentiates inflammasome activation [16].
A randomized, multicenter phase IIa open-label, active comparator trial is underway to assess the efficacy and safety of two regimens of bucillamine compared to colchicine for the treatment of acute gout in patients with moderate to severe gout. Subjects are receiving bucillamine at 900 mg (Arm A) or 1800 mg total dose (Arm B) over 7 days, versus the active comparator colchicine (1.8 mg total dose in 2 doses taken 1 h apart; Arm C) [17]. Results are pending.
In the Pipeline: Agents in Pre-Clinical Development to Target IL-1β or the NLRP3 Inflammasome
Recognition of the centrality of IL-1β and the NLRP3 inflammasome to gouty inflammation has led to other approaches to targeting this pathway for treatment of gouty inflammation. Most of these are still in the pre-clinical phase, and data on their development is generally limited.
-
PENTRA® bodies are small, short-acting (plasma T1/2 ∼1 day) antibody-like fragments of size ∼25 kDa. PENTRA® bodies have excellent tissue penetration and are highly effective in neutralizing their targets (potency in the femtomolar IC50 range (≤600 fM)). Delenex Therapeutics AG is proposing use of a neutralizing IL-1β PENTRA® body for the treatment of acute gout. The high potency and small size may allow IL-1β-directed PENTRA® bodies to have a fast onset and short action in combating acute gouty inflammation [18].
-
Immune Response BioPharma, Inc. is proposing a gout IL-1 β vaccine candidate (Immunereszumab) [19].
-
IL-1 receptor-associated kinase-4 (IRAK-4): A signaling molecule located downstream to IL-1R (Nimbus Therapeutics, LLC) is a kinase protein, long sought after as a target for the treatment of such conditions as systemic lupus erythematosus, rheumatoid arthritis, psoriasis, and inflammatory bowel disease. IRAK-4 inhibition has been shown to block signaling downstream of interleukin-1 receptor and toll-like receptor 4 in vitro and found to be effective in a gout murine peritonitis model [20].
-
CD40 ligand (CD40L): NLRP3 inflammasome activity in macrophages can be terminated through cell to cell contact between the macrophage and effector and/or regulatory T cells. This effect can be mimicked by macrophage stimulation with CD40 ligand (CD40L, CD154), a molecule expressed on the surface of activated T cells. In vitro, a CD40L construct, the adiponectin fusion protein ADIPOQ–CD40L engages CD40 on activated macrophages and shuts off the NLRP3 inflammasome, suppressing IL-1β release and caspase-1 activation [21].
Urate Lowering Therapies: Getting to the Heart of the Problem
While preventing and resolving acute attacks are essential aspects of good gout management, the serum urate level (sU) must also be lowered to <6.0 mg/dl (or lower in some patients) in order to permanently resolve the risk for acute attacks and joint damage [22•]. The first ULTs developed for gout were the uricosuric probenecid (Fig. 2) and the purine analog allopurinol (isomer of hypoxanthine, which inhibits xanthine oxidase (XO) to reduce uric acid production) (Fig. 3) [23•]. Several other uricosurics, including sulfinpyrazone and benzbromarone, have been developed and used outside of the USA, but these carry risks that were deemed unacceptable by the FDA. It was not until 2009 that another medication to lower sU levels in gout patients was approved for use in the USA. Febuxostat is an inhibitor of XO but, in contrast to allopurinol and its metabolites, is minimally excreted through the kidneys [24]. In 2010, a pegylated recombinant uricase, pegloticase, was approved by the FDA for the treatment of hyperuricemia in patients with gout who have failed to normalize sU levels (<6 mg/dl) or continue to have signs and symptoms of gout on standard oral ULT. Pegloticase mechanism of action is unique among current gout therapies, in that it catalyzes the oxidation of uric acid into the more water soluble allantoin, allowing for easy excretion by the kidney [23•].

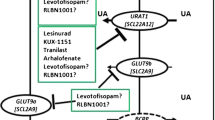
Summary of uric acid handling in the kidney and inhibition by current and pipeline urate lowering therapies. Transporters on the renal tubule epithelial cells contribute to reabsorption of uric acid back into the blood stream or excretion of uric acid in the urine. Modified with permission from Springer from [45]

Uric acid metabolism pathway. Allopurinol, febuxostat, ulodesine, and pegloticase work by decreasing serum urate production as shown above. Modified with permission from Springer from [45]
Lesinurad: Newest Kid on the Block
Most recently, the FDA approved lesinurad, the first selective uric acid resorption inhibitor (SURI). Like probenecid but more potently, lesinurad inhibits urate transporter-1 (URAT1) in renal tubules, thereby promoting uric acid excretion and reducing sU levels. The drug also has lesser uricosuric effects on organic anion transporter 4 (OAT4), a uric acid resorbing transporter that is associated with diuretic-induced hyperuricemia [25•, 26]. Lesinurad is indicated for co-treatment of hyperuricemia with a XO inhibitor in gout patients who have not achieved target sU levels with a XO inhibitor alone.
FDA approval was based on two phase III studies, CLEAR 1 and CLEAR 2 (Combination Study of Lesinurad in Allopurinol Standard of Care Inadequate Responders). CLEAR 1 and CLEAR 2 were replicate 12-month, multicenter, randomized, double-blind, placebo-controlled clinical trials evaluating the efficacy and safety of lesinurad in combination with allopurinol in gout patients with inadequate response to allopurinol [27]. The studies evaluated lesinurad at 200 mg or 400 mg oral, once daily, in combination with allopurinol versus allopurinol plus placebo in subjects with gout aged 18–85 years with sUA ≥6.5 mg/dl at screening [27]. Subjects were required to be on stable doses of allopurinol ≥300 mg (≥200 mg for moderate renal impairment) and have history of ≥2 gout flares in the prior 12 months. The primary endpoint was the proportion of subjects meeting sU target of <6.0 mg/dl by month 6. Secondary endpoints included mean gout flare rate requiring treatment from months 6–12 and proportion of subjects with complete resolution of ≥1 target tophus by month 12.
Both doses of lesinurad in combination with allopurinol increased the proportion achieving sUA target at 6 months by 2–2.5-fold compared to allopurinol alone [27]. As regards adverse effects, lesinurad at the 200 mg dose was comparable to placebo, whereas the 400 mg dose was accompanied in some patients by an increase in serum creatinine levels. Although the majority of serum creatinine increases reversed while on persistent therapy, the drug was approved only at the 200 mg dose level. In contrast to probenecid, lesinurad did not appear to increase the risk of renal stones in either of the dosing groups.
Lesinurad is indicated for hyperuricemia associated with gout in patients who have not achieved target sU levels with a XO inhibitor alone. The drug must be co-administered with a XO inhibitor and is not approved for asymptomatic hyperuricemia. In addition to baseline renal function assessment and periodic monitoring for all patients, more frequent monitoring is required for patients with an estimated CrCl below 60 ml/min. The drug is contraindicated in patients with a CrCl below 30 ml/min and should be discontinued if the CrCl decreases persistently to below 45 ml/min. Lesinurad is also contraindicated for increased sU levels caused by tumor lysis syndrome and Lesch-Nyhan syndrome [28].
Urate Lowering Therapies Under Clinical or Pre-Clinical Study
Despite its ancient provenance, gout continues to be an undertreated disease affecting millions [29•] in part because ULT options have been limited. However, recent advances in our understanding of the pathogenesis of hyperuricemia have promoted continued research leading to the development of novel ULT approaches. Over the past few years, several novel ULT medications for gout that have completed phase I and II studies, and others have entered pre-clinical development. Although it is unlikely that all of the therapies under study will come to FDA approval, the interest and exploration make this an exciting time in the field of gout management.
RDEA3170
RDEA3170 is a novel URAT1 inhibitor found to have a high affinity for URAT1 and greater urate lowering effect compared to other known uricosurics [30]. Phase II, randomized, open-label studies are evaluating the pharmacodynamic effects and safety of RDEA3170 administered in combination with febuxostat, and in combination with allopurinol versus febuxostat and allopurinol monotherapy, respectively. The results of these studies and further clinical trials are pending.
KUX-1151
Pfizer’s KUX-1151 is a new agent that has completed a phase II clinical trial in Japan. KUX-1151 is a potential therapy for gout that has a dual mechanism of action by reducing sU levels through inhibiting both XO and URAT1 [31].
Ulodesine
Ulodesine is a purine nucleoside phosphorylase (PNP) inhibitor with once daily oral dosing that has completed phase II trials [32]. Developed by BioCryst Pharmaceuticals for treatment of gout-associated hyperuricemia, it acts upstream of XO in the purine metabolism pathway to reduce sU production [26]. In doses ranging from 40 to80 mg, once daily ulodesine resulted in lower sU levels in conjunction with allopurinol in patients who had not responded to allopurinol 300 mg daily monotherapy [33]. Another phase II trial tested combination therapy with ulodesine plus various doses of allopurinol and found 100 % of the participants achieved the target of sU less than 6.0 mg/dl [34]. The higher doses of ulodesine led to more diarrhea and rash compared to the placebo group. No increased risk of infection was observed, despite the fact that an inborn error of purine metabolism from PNP deficiency has been associated with a combined immunodeficiency [34]. The future of ulodesine is unclear given there are no current or future studies enrolling patients to date.
Tranilast
Tranilast (Nuon Therapeutics) is approved for bronchial asthma in South Korea and Japan and has inhibitory properties against TGF-β, prostaglandin E2, and IL-1 [35]. It was also found to be an inhibitor of URAT1 in addition to another renal tubular urate pump, GLUT9. In phase II studies, tranilast was shown to significantly lower sU when co-administered with either 400 mg of allopurinol or 40 mg of febuxostat [36, 37]. No further studies have been conducted to date.
RLBN1001
RLBN1001 is a ULT that was stumbled upon by circumstance. Its prototype was found to have urate lowering effects during initial in a study of 350 human subjects being treated for cancer [38]. A proof of concept study in 50 human subjects treated with RLBN1001 found that hypouricemia was associated with increased urinary excretion of both uric acid and oxypurine urate precursors, suggesting bifunctional effects on the production and excretion of urate. Further investigation revealed that RLBN1001 was a potent inhibitor of URAT1 and modest inhibitor of XO, but also a potent clastogen (i.e., inducing disruption or breakage of chromosomes) in an in vitro mouse micronucleus model. Subsequently, the investigators developed a number of analog compounds (RLBN2023, RLBN2024, RLBN3022) without clastogen activity, but still with an inhibitory effect on XO four-fold greater than allopurinol and at least equal to the inhibitory effect of lesinurad on URAT1 [38]. Currently, there are no registered clinical trials pending.
Tofisopam
Tofisopam (Pharmos corporation), a benzodiazepine approved for use outside the USA to treat a variety of disorders associated with stress or autonomic instability, was discovered to have uricosuric properties. Trying to isolate the hypouricemic inducing properties of the compound, investigators found the s-enantiomer of the tofisopam maintained its uricosuric properties without the nervous system effects. Two phase I studies with the s-enantiomer, named levotofisopam, were conducted in the UK and the Netherlands and showed rapid reductions in sU levels [34]. Subsequently, an open-label proof of concept study at the Duke University Clinical Research Unit enrolled 13 subjects, with entry sU levels between 8 and 12 mg/dl [39]. After a 2-week wash out of their current ULT, subjects underwent a 7-day monotherapy dosing period with 50 mg of levotofisopam three times daily. All 13 subjects achieved the target urate level less 6.0 mg/dl, while 77 and 54 % achieved the targets of less than 5.0 mg/dl and less than 4.0 mg/dl, respectively [40]. In the clinical trials, levotofisopam was well tolerated and without unexpected, serious adverse events or significant laboratory abnormalities. To date, no further trials have been registered.
Arhalofenate: Dual ULT/Anti-Inflammatory Therapy
Patients initiating ULTs enter into a period of increased vulnerability to acute gout attacks, presumed to relate to the release of pro-inflammatory crystals during the disruption of deposited urate aggregates. On this basis, guidelines recommend that all patients starting ULT also initiate daily anti-inflammatory prophylaxis for no less than 6 months. An agent that could both lower sU, and serve as a daily anti-inflammatory, might increase patient ease and compliance through the simplification of the treatment regimen.
Arhalofenate, developed as a peroxisome proliferator-activated receptor gamma (PPAR-γ) partial agonist, was serendipitously discovered to have urate lowering properties during clinical trials for diabetes [34]. Decreases in sU were found to be due to blockade of the transporters URAT-1, OAT4, and OAT10, preventing the reabsorption of uric acid in the renal tubes and allowing for increased sU renal clearance. Additionally, arhalofenate was found to inhibit upregulation of IL-1β, thereby providing simultaneous ULT and prophylactic anti-inflammatory effects. A randomized, double-blind, placebo-controlled phase IIb trial at 54 centers involving 239 gout patients was recently completed to assess arhalofenate safety and efficacy in both controlling inflammation and lowering sU [41]. Subjects were randomized 1:2:2:2:2 to placebo, arhalofenate 600 mg or 800 mg, allopurinol 300 mg, or allopurinol 300 mg combined with colchicine 0.6 mg dose once daily for 12 weeks. The primary outcome comparing flare rates between the arhalofenate 800 mg to allopurinol 300 mg and allopurinol 300 mg plus colchicine was met with a 46 % improvement in the arhalofenate group (p = .0056). Neither the 600 mg nor the 800 mg dose of arhalofenate showed a greater sU lowering efficacy than 300 mg of allopurinol without or with colchicine, but both were significantly more effective than placebo (−1, −12, 16, −29, −25 % change, respectively) [41]. Further trials are pending [34].
Conclusions
Despite our evolving understanding of how hyperuricemia and gout occur, it still continues to be a commonly undertreated disease. Lack of patient and provider education, failure to treat to target, underdosing, and contraindications (or perceived contraindications) to available therapies all contribute to many patients continuing to struggle with gouty arthritis. In some cases—particularly in patients with more severe gout and/or higher urate levels—currently available treatments may be limited in their ability to achieve the needed target sU. Additionally, all of the oral agents currently used to treat acute gout attacks may require caution in the setting of comorbid conditions that commonly accompany the diagnosis of gout. Therefore, future medications for acute gout should more effectively target specific steps of the inflammatory cascade while limiting unwanted and potentially dangerous adverse effects. The amelioration of gouty inflammation using IL-1-inhibiting biological therapies, and other compounds directed at NLRP3 inflammasome activation or function, is an exciting example of biological understanding leading to targeted therapeutics. The possibility of small molecule therapies that offer less expensive and more convenient approaches to Il-1 inhibition would be attractive alternatives to current parenteral IL-1-targeting agents.
The potential advantages of more potent and better tolerated anti-inflammatory therapies include not only decreased pain and shortened disability during an acute attack, but also better compliance and a greater window to reach target sU levels. More specific anti-inflammatory therapies such as the IL-1 inhibitors may effectively treat and prevent acute attacks, without affecting co-existing diabetes, hypertension, and renal disease. More effective and/or rapidly acting ULTs, that help people achieve target in a shorter period time, may provide an opportunity to decrease overall morbidity for patient as well as healthcare cost savings in the long run. Dual ULT/anti-inflammatory therapies may simplify patient regimens and improve compliance. Although there are a limited number of studies evaluating the cost effectiveness of new versus old gout therapies, there is some evidence that dose escalation and changing therapy (from old to new) with a more aggressive target may be cost effective when taking into account quality of life and disability of life years [42••, 43••]. Additionally, new treatments can be an impetus for improving gout education of providers and patients, provider to provider co-management, and subsequent opportunities to improve outcomes and establish new best practices.
Some promising ULTs for gout, such as tranilast, ulodesine, and KUX-1151, may have been shelved or stalled in development. But others such as the uricosurics RDEA3170, or the dual action drug arhalofenate, provide optimism for the future of gout management and the patients it afflicts. While there have been just three new medications to lower sU levels over the last decade, the revitalization of the discussion, education, and research around this most common of inflammatory arthropathies continues to gain momentum. In addition to a new staging system that has been proposed that goes beyond the traditional symptomology stages designated as hyperuricemia, acute intermittent, and advanced gout [44], new guidelines have been introduced across the globe regarding the management of gout. With every new gout therapy developed, the bar in how gout is managed is raised a little higher, and one day, we may be able to say that gout is a curable disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
http://store.globaldata.com/market-reports/pharmaceuticals-and-healthcare/opportunityanalyzer-gout-opportunity-analysis-and-forecast-to-2018#.Vso-ZVJflfQ. Accessed 2/21/16.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41. This study demonstrates the effect of monosodium urate crystals on the NALP3 inflammasome.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9(2):R28.
Chen K, Fields T, Mancuso CA, Bass AR, Vasanth L. Anakinra’s efficacy is variable in refractory gout: report of ten cases. Semin Arthritis Rheum. 2010;40(3):210–4.
Ottaviani S, Molto A, Ea HK, Neveu S, Gill G, Brunier L, et al. Efficacy of anakinra in gouty arthritis: a retrospective study of 40 cases. Arthritis Res Ther. 2013;15(5):R123.
Schlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis. 2012;71(11):1839–48. This study was the basis for the approval of the IL- β inhibitor Canakinumab in gout by the European Medicine Agency.
Schlesinger N, Mysler E, Lin HY, De Meulemeester M, Rovensky J, Arulmani U, et al. Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis. 2011;70(7):1264–71.
European Medicine Agency: Ilaris http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001109/human_med_000826.jsp&mid=WC0b01ac058001d124. Accesses 2/20//16.
FDA Arthritis Advisory Committee Meeting June 21, 2011: Supplemental Biologic License Application (sBLA) 125319/25 Canakinumab (Ilaris®) for the Treatment of Gouty Arthritis Attacks http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/arthritisadvisorycommittee/ucm263001.pdf. Accessed 2/20/16.
Khanna D, Khanna PP, Fitzgerald JD, Singh MK, Bae S, Neogi T, et al. 2012 American college of rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64(10):1447–61. Part 2 of the treatment guidelines for gout published by the ACR.
Nanke Y, Iwatani M, Kobashigawa T, Yago T, Yamanaka H, Kotake S. Radiographic repair in three Japanese patients with rheumatoid arthritis treated with bucillamine. Mod Rheumatol Jpn Rheum Assoc. 2009;19(6):681–6.
Amersi F, Nelson SK, Shen XD, Kato H, Melinek J, Kupiec-Weglinski JW, et al. Bucillamine, a thiol antioxidant, prevents transplantation-associated reperfusion injury. Proc Natl Acad Sci U S A. 2002;99(13):8915–20.
Whitekus MJ, Li N, Zhang M, Wang M, Horwitz MA, Nelson SK, et al. Thiol antioxidants inhibit the adjuvant effects of aerosolized diesel exhaust particles in a murine model for ovalbumin sensitization. J Immunol. 2002;168(5):2560–7.
Wielandt AM, Vollrath V, Farias M, Chianale J. Bucillamine induces glutathione biosynthesis via activation of the transcription factor Nrf2. Biochem Pharmacol. 2006;72(4):455–62.
Tsuji F, Miyake Y, Aono H, Kawashima Y, Mita S. Effects of bucillamine and N-acetyl-L-cysteine on cytokine production and collagen-induced arthritis (CIA). Clin Exp Immunol. 1999;115(1):26–31.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40.
Bucillamine for the Treatment of Acute Gout Flare in Subjects With Moderate to Severe Gout. Clinal Trials.Gov https://www.clinicaltrials.gov/ct2/show/NCT02330796?term=gout&recr=Open&rank=9. Accessed 2/21/16.
http://www.finanzwire.com/finance/stocks/delenex-therapeutics-ag/news/353519.html. Accessed 2/21/16.
http://www.immuneresponsebiopharma.com/Pages/Immunereszumab.aspx. Accessed 2/21/16.
Bree APK, Benson M, Dower K, Shen M, Lee K. Pharmacological inhibition of interleukin-1 receptor-associated kinase-4 reduces inflammation in a murine model of gout and is consistent with IL-1 signaling blockade. Arthritis Rheum. 2011;63 Suppl 10:1621.
Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460(7252):269–73.
Khanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, et al. 2012 American college of rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64(10):1431–46. Part 1 of the treatment guidelines for gout published by the ACR.
Keenan RT. Safety of urate-lowering therapies: managing the risks to gain the benefits. Rheum Dis Clin N Am. 2012;38(4):663–80. A thorough review of traditional urate lowering therapies as well as potential side effects and contraindications one may encounter with gout patients.
Keenan RT, Pillinger MH. Febuxostat: a new agent for lowering serum urate. Drugs Today (Barc). 2009;45(4):247–60.
Fleischmann R, Kerr B, Yeh LT, Suster M, Shen Z, Polvent E, et al. Pharmacodynamic, pharmacokinetic and tolerability evaluation of concomitant administration of lesinurad and febuxostat in gout patients with hyperuricaemia. Rheumatology (Oxford). 2014;53(12):2167–74. A recently published phase 1B trial of the recently approved selective uricosuric lesinurad.
Crittenden DB, Pillinger MH. New therapies for gout. Annu Rev Med. 2013;64:325–37.
Lesinurad, a Novel Selective Uric Acid Reabsorption Inhibitor, in Two Phase III Clinical Trials: Combination Study of Lesinurad in Allopurinol Standard of Care Inadequate Responders (CLEAR 1 and 2) [http://acrabstracts.org/abstract/lesinurad-a-novel-selective-uric-acid-reabsorption-inhibitor-in-two-phase-iii-clinical-trials-combination-study-of-lesinurad-in-allopurinol-standard-of-care-inadequate-responders-clear-1-and-2/. Accessed 2/21/16.
Zurampic Package Insert http://www.azpicentral.com/zurampic/zurampic.pdf. Accessed 2/21/16.
Keenan RT, O’Brien WR, Lee KH, Crittenden DB, Fisher MC, Goldfarb DS, et al. Prevalence of contraindications and prescription of pharmacologic therapies for gout. Am J Med. 2011;124(2):155–63. This study of a large gout cohort demonstrates the multiple comorbidities and contraindications to traditional gout therapies that patients and providers face when making treatment decisions.
Miner J TP: RDEA3170 a novel high affinity URAT1 inhibitor binds to central domain within URAT1. Ann Rheum Dis. 2012; 71(Suppl 3): p 446. Ann Rheum Dis. 2013; 71(Suppl 3):446.
Phase II Exploratory Clinical Study of KUX-1151 [https://clinicaltrials.gov/ct2/show/study/NCT02190786].
Study to Evaluate sUA Lowering Activity, Safety and Efficacy of Oral Ulodesine Added to Allopurinol [https://clinicaltrials.gov/ct2/show/NCT01265264?term=ulodesine&rank=2].
Hollister AS BM, Terkeltaub R, Waugh A, Lyman S, Flynt A, Fitz-Patrick D. BCX4208 synergistically lowers serum uric acid (sUA) levels when combined with allopurinol in patients with gout: results of a phase 2 dose-ranging trial. Arthritis Rheum. 2011;63(10 (Suppl)):1018.
Edwards NL, So A. Emerging therapies for gout. Rheum Dis Clin N Am. 2014;40(2):375–87.
Suzawa H, Kikuchi S, Ichikawa K, Koda A. Inhibitory action of tranilast, an anti-allergic drug, on the release of cytokines and PGE2 from human monocytes-macrophages. Jpn J Pharmacol. 1992;60(2):85–90.
Study of Tranilast Alone or in Combination With Allopurinol in Subjects With Hyperuricemia [https://clinicaltrials.gov/ct2/show/NCT01052987?term=tranilast&rank=2].
Study of tranilast alone or in combination with febuxostat in patients with hyperuricemia [https://clinicaltrials.gov/ct2/show/NCT00995618?term=tranilast&rank=4].
Warrell RPKA, Barnes K, Satyanarayana C. Profound hypouricemia induced in human subjects by novel bifunctional inhibitors of xanthine oxidase and URAT1. Arthritis Rheum. 2014;66(11 (Suppl)):S366.
Study of Levotofisopam 50 mg Three Times a Day (TID) Administered for 7 Days on Hyperuricemia and Gout [https://clinicaltrials.gov/ct2/show/NCT01519687?term=levotofisopam&rank=1].
Noveck RJ W, Z, Forsthoefel A, Sigmon K, Hall PC, Keogh JC, Sundy JS. Levotofisopam has uricosuric activity and reduces serum urate levels in patients with gout. Arthritis Rheum. 2012; 64:(Suppl 10).
Steinberg A CH, Choi YJ, Martin R, McWherter C, Zhang Y, Boudes P. A Study to evaluate the efficacy and safety of arhalofenate for preventing flares and reducing serum uric acid in gout patients [abstract]. Arthritis Rheum. 2015; 67 (suppl 10).
Beard SM, von Scheele BG, Nuki G, Pearson IV. Cost-effectiveness of febuxostat in chronic gout. Eur J Health Econ HEPAC Health Econ Prev Care. 2014;15(5):453–63. This study showed febuxostat as cost-effective and suitable second-line option for ULT for the treatment of gout patients (including a history or presence of tophus and/or gouty arthritis) when treatment with allopurinol was inadequate, not tolerated, or contraindicated.
Jutkowitz E, Choi HK, Pizzi LT, Kuntz KM. Cost-effectiveness of allopurinol and febuxostat for the management of gout. Ann Intern Med. 2014;161(9):617–26. This study showed the cost-saving when treating with allopurinol a single therapy compared with no treatment, and the cost effectiveness of allopurinol then febuxostat sequential therapy.
Dalbeth N, Stamp L. Hyperuricaemia and gout: time for a new staging system? Ann Rheum Dis. 2014;73(9):1598–600.
Mohammad S, Giattino SL, Keenan RT. Current and emerging therapies for gout. Curr Treatm Opt Rheumatol. 2015;1(2):143–55.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
RTK reports personal fees from Astra Zeneca, personal fees from Takeda, grants from Novartis, personal fees from Savient, and personal fees from Crealta, outside the submitted work. NS reports grants and personal fees from Astra Zeneca, personal fees from Pfizer, personal fees from Celgene, grants and personal fees from Novartis, personal fees from Takeda, personal fees from Sobi, consulting paid to institution for BMS, personal fees from Alkermes, and consulting paid to institution from SGS North America, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Crystal Arthritis
Rights and permissions
About this article

Cite this article
Keenan, R.T., Schlesinger, N. New and Pipeline Drugs for Gout. Curr Rheumatol Rep 18, 32 (2016). https://doi.org/10.1007/s11926-016-0579-7
Published:
DOI: https://doi.org/10.1007/s11926-016-0579-7