
Abstract
Purpose of Review
Inflammasomes are multimeric protein structures with crucial roles in host responses against infections and injuries. The importance of inflammasome activation goes beyond host defense as a dysregulated inflammasome and subsequent secretion of IL-1 family members is believed to be involved in the pathogenesis of various diseases, some of which also produce skeletal manifestations. The purpose of this review is to summarize recent developments in the understanding of inflammasome regulation and IL-1 family members in bone physiology and pathology and current therapeutics will be discussed.
Recent Findings
Small animal models have been vital to help understand how the inflammasome regulates bone dynamics. Animal models with gain or loss of function in various inflammasome components or IL-1 family signaling have illustrated how these systems can impact numerous bone pathologies and have been utilized to test new inflammasome therapeutics.
Summary
It is increasingly clear that a tightly regulated inflammasome is required not only for host defense but for skeletal homeostasis, as a dysregulated inflammasome is linked to diseases of pathological bone accrual and loss. Given the complexities of inflammasome activation and redundancies in IL-1 activation and secretion, targeting these pathways is at times challenging. Ongoing research into inflammasome-mediated mechanisms will allow the development of new therapeutics for inflammasome/IL-1 diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Inflammasome and IL-1 Family
The innate immune system provides the first line of defense against pathogens due to its ability to recognize a wide range of pathogens through germline-encoded receptors called pattern recognition receptors (PRRs). PRRs are expressed by numerous myeloid cell types including macrophages, monocytes, osteoclasts, dendritic cells, neutrophils, as well as non-myeloid cell types such as epithelial, endothelial, and mesenchymal cells. When activated, PRRs rapidly trigger downstream signaling pathways which lead to inflammatory responses. PRRs recognize conserved microbial molecular structures termed pathogen-associated molecular patterns (PAMPs) as well as danger-associated molecular patterns (DAMPs) released by injured cells or tissues.
The interleukin-1 (IL-1) family of cytokines is primarily associated with innate immunity and affects a broad spectrum of inflammatory and immune responses. Certain members of the IL-1 family (IL-1β and IL-18) require caspase-mediated proteolytic cleavage for functional activation [1]. The inflammasome is a macromolecular structure that assembles in the cytosol of cells in response to infection (via PAMPs) or tissue injury (via DAMPs) and leads to the activation of inflammatory caspases (caspase-1 and -11 in mouse, and -1, -4, and -5 in humans) [2]. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 into their biologically active forms [3, 4]. Gasdermin D (GSDMD) is also cleaved and activated by inflammatory caspases, which leads to the assembly of GSDMD pores in the plasma membrane, enabling the release of active IL-1β and IL-18 into the extracellular milieu [5].
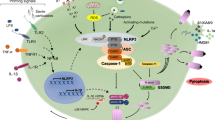
Multiple types of inflammasomes have been identified [3, 6], and can be divided into two broad categories: (1) canonical, which activate caspase-1, and (2) noncanonical, which activate caspase-11 in mouse and caspase-4/5 in human. Canonical inflammasome signaling first requires a licensing step (also known as priming), in which cytokines (e.g., TNF, IL-1) or PAMPs upregulate the expression of pro-IL-1β, and in some cases the inflammasome sensor molecule (e.g., NLRP3) (Fig. 1). Inflammasome assembly requires a second signal (e.g., DAMP recognition by an inflammasome sensor), which results in inflammasome sensor activation and oligomerisation. Next, there is recruitment of the inflammasome adaptor protein (e.g., ASC; apoptosis-associated speck-like protein containing a CARD (caspase recruitment domain), encoded by the Pycard gene) (Fig. 1). Once the sensor protein oligomerizes and interacts with ASC, they form a single macroscopic speck in the cytosol. This speck subsequently docks pro-caspase-1 monomers triggering caspase-1 activation by dimerization and autocatalysis [4] (Fig. 1). Active caspase-1 then cleaves pro-IL-1β and pro-IL-18 into biologically active cytokines. The final step is the release of mature IL-1β and IL-18 from the cytosol into the extracellular fluid, which occurs as a consequence of passage of these cytokines through GSDMD pores across the plasma membrane [7], and drives a particular form of cell death called pyroptosis (Fig. 1). In non-canonical inflammasome signaling, caspases are able to directly bind intracellular lipopolysaccharide from Gram-negative bacteria and can directly mediate GSDMD cleavage and pyroptosis, as well as trigger a secondary activation of the canonical inflammasome [8].

Assembly of the NLRP3 inflammasome. Following a priming signal, e.g., recognition of a PAMPs by PRRs (1) the transcription of Nlrp3 and Il1b genes are activated in a NF-κB-dependent manner (2). In parallel, an activation signal is provided by a variety of stimuli (DAMPs, e.g., ATP, K+ ionophores) (3), all of which lead to activation of the NLRP3 sensor protein, NLRP3 oligomerization and subsequent association with the ASC adaptor subunit, which forms a large ASC-containing speck with subsequent docking of pro-caspase-1 (4), where pro-caspase-1 dimerization and auto-catalysis produces active caspase-1 (5), which then cleaves and activates pro-IL1β, pro-IL-18 and GSDMD (6). Cleaved GSDMD forms pores within the membrane which enables release of mature IL-1β and IL-18 from the cell and triggers pyroptosis (6). Other canonical inflammasomes function in a similar manner but with different sensor proteins. Created with BioRender.com
Not all IL-1 family members require cleavage via inflammatory caspases. For example, IL-1α is constitutively expressed as pro-IL-1α, which is biologically active but can be further activated by inflammasome-independent proteolytic cleavage. Inflammatory stimuli also enhance the transcription of the Il1a gene. Pro-IL1α binds to the plasma membrane of phagocytes and can be further activated and released via protease-mediated cleavage by chymase, granzyme B, or neutrophil elastase, or be released in a soluble form [9]. Pro-IL-33 and pro-IL-37 are also biologically active [10]. A hallmark of IL-1 signaling is the redundancy of IL-1 family members capable of binding the same cognate receptor. For example, IL1R1 binds IL-1α, IL-1β, and their endogenous antagonist IL-1RA albeit with different affinities [11]. IL-1 signaling can be controlled via the endogenous IL-1 antagonist IL1RA, that competes for the receptor as a negative feedback inhibitor of excessive IL-1 signaling [1].
IL-18 forms a complex with the IL-18 receptor α and β chain (IL-18Rα and IL-18Rβ) which activates downstream signaling. IL-18 signaling is regulated by IL-18 binding protein (IL-18 BP), a circulating endogenous IL-18 antagonist with high affinity for IL-18 [12]. IL-37 exerts an anti-inflammatory effect by binding to IL-18Rα, but recruits IL-1R8 instead of IL-18 Rβ [13].
It is clear that inflammasome signaling is vital for rapid responses to infection and injury; however, inflammasome dysregulation and aberrant IL-1 expression also drives the initiation and progression of arthrosclerosis [14], type 2 diabetes and obesity [15], neurodegenerative diseases [16], and cancer [17]. In this review, we further discuss functions for inflammasomes and IL-1 family activity in bone homeostasis as well as in pathologies of bone accrual and loss, as well as current and future therapies for these diseases.
Inflammasomes and IL-1 in Physiological Bone Development
It is well established that the skeletal system can be impacted by chronic inflammation but less appreciated is the role that the inflammasome and IL-1 plays in physiological bone development.
Primary murine osteoblasts express NLRP3, ASC, and caspase-1 mRNA and proteins [18]. Osteoblasts present on bone surfaces also express NLRP3 protein [19]. While numerous commercially available Nlrp3−/− strains do not report bone phenotypes, one study established in 4-week-old male and female Nlrp3−/− mice display impaired skeletal development with reduced full body length, lower trabecular bone volume, lower trabecular thickness and number, as well as a deficient growth plate [19]. NLRP3 expression has also been observed in hypertrophic chondrocytes, and Nlrp3−/− mice display reduced bone sialoprotein expression in hypertrophic chondrocytes [19], suggestive of a role for NLRP3 during chondrocyte maturation. In vitro generated osteoclasts also express NLRP3 and NLRC4 (NLR CARD domain containing-4, an adaptor for the NLRC4 inflammasome) when exposed to LPS and nigericin [20] and the loss of NLRP3 or NLRC4 inflammasomes in mice attenuated osteoclast differentiation in vitro. Interestingly, this study by Alippe and colleagues observed that male Nlrp3−/− mice (but not Nlrc4−/−) at 8 months of age, displayed a significantly higher bone mass and bone mineral density compared to wild-type counterparts [20]. This observation is in stark contrast to the study by Detzen at al. mentioned above [19]. These contrasting conclusions highlight the complexities of using small animal models where analysis of bone phenotypes at different ages (4 weeks versus 8 months) or different sex may potentially provide explanations for these phenotypic discrepancies.
IL-1 was originally identified as ‘osteoclast-activating factor.’ IL-1 has been proposed to stimulate osteoclastogenesis under physiological conditions indirectly by stimulating prostaglandin E2 synthesis in osteoblasts [21] as well as increasing the expression of receptor activator of NF-κB ligand (RANKL) expression in osteoblasts [22]. In vitro studies also confirmed that IL-1 directly promotes osteoclast formation, multinucleation, pit forming activity [23], as well as survival [24, 25]. Mice administered IL-1β develop hypercalcemia and display increased osteoclast numbers, and treatment with chimeric osteoprotegerin (OPG; a RANKL antagonist) reduced hypercalcemia and osteoclast number [26]. IL-18 is expressed by osteoblasts [27] and chondrocytes [28], IL-18 also inhibits osteoclastic bone resorption indirectly [27] and upregulates OPG in stromal/osteoblast cells [29]. Small animal models have provided further insights into how the IL-1 family cytokines influence bone development. Under germ-free conditions, Il1a−/−, Il1b−/−, or Il1a−/− x Il1b−/− mice have increased bone mass (femur mineral density, trabecular bone mass, and cortical thickness) compared to wild-type controls [22], suggestive of roles in physiological bone metabolism. In addition, mice defective for the Il1r1 gene exhibit a narrow growth plate, due to a smaller hypertrophic zone [30], suggestive of a role in hypertrophic chondrocyte differentiation. However, these observations are in contrast to another study finding no changes in bone mass in Il1r1−/− mice, concluding that normal bone development can occur in the absence of IL-1R1 [31]. These contrasting conclusions highlight the complexities of small animal models with differences in mouse genetic backgrounds, housing conditions, and bone mass measurement methods all being suggested as the reasons behind these discrepancies. Finally, transgenic mice that overexpress IL-18 were shown to have decreased trabecular bone turnover and altered cortical structure [32]; however, no bone phenotype has been reported for Il18−/− mice.
Overall, a few studies have linked the inflammasome and IL-1 family members to bone development, but to date, most research has focused on inflammasome regulation for prevention of disease, which is further discussed in sections below.
Inflammasomopathies with Skeletal Phenotypes
The term ‘inflammasomopathies’ describes a group of mechanistically similar diseases which have inappropriate inflammasome activation. The diseases with impacts on the skeleton are further discussed below.
Cryopyrin-Associated Periodic Syndromes
Cryopyrin-associated periodic syndromes (CAPS) are autoinflammatory disorders mediated by dysfunctional inflammasome activation caused by heterozygous germline or somatic gain-of-function mutations in the NLRP3 gene [33, 34]. These mutations lead to a hyperactive NLRP3 inflammasome, increased activation, and cleavage of caspase-1 and subsequent IL-1β and IL-18 cleavage, activation, and secretion. Clinically, CAPS refers to three disorders (listed in order of increasing disease severity): familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID), all with shared and unique clinical features. CAPS diagnosis can be challenging, as patients show a heterogeneous multi-system clinical presentation. CAPS is frequently characterized by cutaneous, neurological, ocular, and otologic features. Numerous studies have also established that CAPS patients display varying (20–90%) levels of musculoskeletal manifestations such as arthralgia, arthritis, and myalgia, with more severe skeletal abnormalities such as patellar overgrowth, bone deformity, bone/joint erosion, and/or osteolytic lesions [35,36,37] (Table 1). NOMID is the most severe form of CAPS. NOMID arthropathy begins in infancy [33] with increased patellar and epiphyseal long bone ossification, and development of calcified masses that deform the adjacent metaphysis and epiphysis [35]. These skeletal deformities are a common cause of disability in NOMID patients. NLRP3 is not only expressed in monocytes and neutrophils but also in human chondrocytes, accounting for the arthropathy seen in these syndromes [34].
In order to further understand CAPS disease pathogenesis, gain-of-function small animal models were developed [40,41,42]. One group developed two models, each with Cre recombinase inducible mutations Nlrp3A350V and Nlrp3L351P, corresponding to the human mutations NLRP3A352V and NLRP3L353P [41], which are associated with MWS and FCAS. A NOMID mouse model was also developed which contains a knock-in for the Nlrp3D301N mutation (ortholog of NLRP3D303N in humans), a mutation associated with NOMID patients [40]. These mouse models replicate clinical and pathologic features seen in humans and have arthropathy such as growth retardation and osteopenia; however, both models demonstrated early mortality. These models have confirmed a pivotal, but not exclusive, role for IL-1β in CAPS [41], whereby treatment with the small synthetic NLRP3 inflammasome inhibitor MCC950 effectively and specifically blocked NLRP3 activation in vivo with increased survival and body weight [43]. Neutrophils have been suggested as a cellular driver of CAPS pathology [44]. Gasdermin D deficiency has also been demonstrated to reduce NOMID pathogenesis in mice [45•], which supports discovery efforts aimed at identifying selective gasdermin D inhibitors.
IL-1 blockade has been the standard of care for CAPS patients. Specifically, Anakinra (a recombinant human IL-1 receptor antagonist) [46], Rilonacept (a recombinant fusion protein consisting of the extracellular domain of IL-1 receptor fused with IgG1 Fc region; IL-1 trap) [47], and Canakinumab (a humanized neutralizing IL-1β monoclonal antibody) have all been trialed [48]. Treatment resulted in reduced inflammatory symptoms such as rash severity, conjunctivitis, as well as reduced C-reactive protein (CRP) and serum amyloid A (SAA) levels, and over time, reduced organ inflammation [49, 50]. However, a consensus has not been reached on whether IL-1 antagonist therapies are routinely successful in reducing CAPS skeletal manifestations. There are studies that did not report positive outcomes [51]. However, there are a few studies indicating that IL-1 antagonist therapies did result in beneficial skeletal outcomes but these are only in a few patients. For example, one study concluded that anakinra treatment (1 mg/kg/day) had no influence on clinical manifestations not closely related to ‘inflammation’ such as bone dysplasia. In this study, 4 patients with growth failure present at baseline showed no growth catch up after treatment. However, in one patient, there was a dramatic clinical and radiologic improvement in a chodroblastoma after 2 years of anakinra [52]. An additional study, similarly, showed an improvement in arthropathy in one NOMID infant after anakinra treatment. Radiological examination confirmed metaphyseal fraying and cupping and widening of the growth plate in the distal femur at baseline, with follow-up radiographs indicating a reduction in growth plate widening and longitudinal femur and tibia growth after 20 months of observation [53]. Another group reported CAPS patients at baseline, displayed subnormal BMD values (mean (SD) Z-scores of − 2.22 (0.51), − 1.96 (1.39), − 1.92 (1.92), and − 1.75 (1.84) at the femoral neck, L1–L4, total hip and intertrochanteric, respectively) and after 60 months of anakinra the mean Z scores increased (− 1.08 (1.63), − 0.74 (1.47), − 0.80 (1.60), and − 0.96 (1.63) at the femoral neck, L1–L4, total hip, and intertrochanteric, respectively) [54]. More recently, a longer-term study was able to illustrate some resolution of arthropathy after anakinra in some but again not all patients [55]. Clearly, larger studies are necessary to further assess whether IL-1 antagonist therapies provide significant improvement in the bone pathologies observed in CAPS patients.
CAPS patients also display elevated levels of ASC and mature IL-1β in peripheral blood mononuclear cells and treatment of mononuclear cells ex vivo with MCC950 reduced mature IL-1β production, highlighting the therapeutic potential of NLRP3 small molecule inhibitors [56•]. As deubiquitination is essential for efficient NLRP3 inflammasome activity, blocking deubiquitination prevented IL-1β production and inhibited the activation of multiple mouse NLRP3 mutants linked with CAPS [57•], which were previously shown not to benefit from MCC950 treatment [58]. This also suggests that posttranslational modification of NLRP3 is an additional future pharmacological target for CAPS.
Other treatments such as non-steroidal anti-inflammatory drugs, antihistamines, and immunosuppressants were trialed with disappointing results [59].
Deficiency in IL-1 Receptor Antagonist
Deficiency in IL-1 receptor antagonist (DIRA) is a rare autosomal recessive autoinflammatory disease in which a deletion in the IL1RN gene (which encodes IL-1RA) results in a loss of production of IL-1RA [60]. As IL-1RA is a natural endogenous inhibitor of IL-1 [61], DIRA infants have unopposed stimulation of IL1R1 [60]. DIRA has disease phenotypes similar to CAPS, where neonates present with skin and bone manifestations. Over time, DIRA patients present with an array of skeletal abnormalities such as ballooning of long bones and anterior rib ends, multifocal osteolytic lesions, widening of clavicles, metaphyseal erosions, and heterotopic ossifications at the proximal end of femurs [60, 62]. DIRA has been studied in small animal models using mice defective for IL-1RA or in IL-IRA overexpression models, and similar to humans, mice that lack IL-1RA display arthritis and skin manifestations [63,64,65]. However, development of some arthropathy, such as bone erosions, was dependent the background strain of mice [63,64,65].
Similar to CAPS, DIRA patients are routinely administered with the IL-1R antagonist anakinra. Treatment has shown to rapidly resolves all signs of DIRA including severe osteopenia and bone lesions [66].
Pathological Roles of IL-1 in the Skeleton
It is increasingly clear that dysregulated inflammasome action and subsequent IL-1 secretion have pathological effects on the skeleton (Table 2). Herein, we outline the pathologies associated with aberrant inflammasome/IL-1 family actions with pathologies of bone accrual and loss.
Bone Accrual Associated Diseases
Heterotopic Ossifications
Neurogenic heterotopic ossifications (NHO) are bones that develop in periarticular muscles and are a frequent complication after spinal cord injuries (SCI). NHOs frequently lead to ankylosis of the affected joints [93], and cause nerve and blood vessel compression, with treatment limited to surgical resection [94, 95]. Using a mouse model of NHO after SCI, we have established that IL-1 signaling contributes to NHO pathology in mouse and humans [67•]. Il1r1−/− mice had significantly reduced NHO development compared to wild-type controls, and IL-1β was significantly enhanced in the plasma of NHO patients and expressed by CD68+ macrophages in human NHO biopsies. As previously mentioned, a high proportion of infants with DIRA (unopposed IL-1 signaling) develop periarticular heterotopic ossifications [62]. Given all these observations, we investigated the role of the NLRP3 inflammasome in NHO development. Nlrp3 and Pycard mRNA was significantly upregulated after muscle injury, irrespective of SCI (Fig. 2a, b). Interestingly, there was no change in NHO bone volumes in mice administered MCC950 or in mice defective for both Casp1 [96] and Casp11 genes (ICE−/−) compared to controls (Fig. 2c, d). Given that NHO bone volumes were not attenuated in Il1a−/− or Il1b−/− mice [67•], or when components of the inflammasome were inhibited or absent, both our studies suggests that both IL-1 proteins act in concert and specifically targeting the NLRP3 inflammasome or either of the two IL-1 genes separately, is not sufficient to reduce NHO development.

The Inflammasome and NHO pathogenesis. a, b C57BL/6 mice underwent spinal cord injury (SCI) surgery and muscle injury was induced via an intramuscular injection of CDTX or control PBS injection, or SHAM surgery with either an intramuscular injection of CDTX or PBS injection. Both Nlrp3 (a) (**p = 0.0012, p = 0.0015) and Pycard (b) (**p = 0.0096, ****p < 0.0001) mRNA expression was upregulated in whole muscle by CDTX intramuscular injection on day 4 post-injury. c C57BL/6 mice underwent SCI with an intramuscular injection of CDTX and treated with either PBS or MCC950 (20 mg/kg daily ip) for the first 7 days post-surgery. NHO volumes measured by microcomputed tomography (μCT), with two representative images per treatment group. d C57BL/6 and ICE−/− mice underwent SCI with an intramuscular injection of CDTX, NHO volumes measured by μCT, with two representative images per group. Each dot represents one mouse; results are presented as mean ± SD, single experiments
There is also evidence of IL-1-driven pathogenesis in other forms of heterotopic ossification (HO). Increased plasma IL-1β was also noted in an Achilles’ tenotomy and dorsal burn injury mouse model of trauma-induced HO [97]. Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal genetic disease caused by dominant gain-of-function mutations in the coding sequence of the ACVR1 gene, encoding the activin A receptor type 1, a bone morphogenetic protein (BMP) type I receptor [98]. FOP children develop soft tissue swellings early in life and develop progressive HO which cover the whole body becoming ultimately fatal. In a BMP-induced mouse model of HO, anti-IL-1 therapy abolished HO development [68]. In the Acvr1R206H mouse model of FOP, IL-1β was detected by immunohistochemistry at higher levels in Acvr1R206H/+ lesions compared to wildtype [99]. Anakinra has also been administered to FOP patients. During ‘flare-ups,’ IL-1β plasma levels increased, which reduced after anakinra [100]; however, there were insufficient data to determine how important IL-1 therapy was in comparison to therapies targeting other inflammatory cascades.
Fracture and Bone Defect Healing
Diabetics are known to have increased fracture risk and impaired bone healing, and the inflammasome has been shown to play a role in fracture repair in this context. In a mouse model of diabetic-induced fracture, treatment with the NLRP3 inflammasome inhibitor glyburide reduced pro-inflammatory cytokine expression, fracture-associated osteoclasts and also increased callus volume and maximum torque and yield torque [69]. Targeting NLPR3 via short hairpin RNA also improved alveolar bone defect healing in diabetic rats [70].
In other non-diabetic fracture models, IL-1β was upregulated during early phases of bone repair [71], and a role for IL-1β in hematoma fibrin clots has been proposed [101]. IL-1β administration in a rat tibial bone defect model significantly increased osteoblasts number at the defect site [102]; however, there was no change in callus size, cartilage volume, and bone volume in Il1r1−/− mice [71]. Finally, repeated doses of IL-1β after fracture had no impact on healing, suggesting a minor or a at least a redundant role for IL-1 in non-diabetic fracture healing.
Ankylosing Spondylitis
Ankylosing spondylitis (AS) is a chronic autoinflammatory disease affecting the axial skeleton including the spine and sacroiliac joints, where between 1 and 33% of patients develop syndesmophytes (bony spurs occurring at vertebral corners that potentially fuse the spine) over 2 years of disease progression [103].
Peripheral blood mononuclear cells (PBMC) isolated from AS patients express higher mRNA levels of NLRP3, PYCARD, NLRC4, CASP1, and increased serum IL-1β and IL-18 compared to healthy controls [104]. To date, only a few clinical studies on a small number of patients have evaluated the effects of anakinra in managing inflammation in AS. In one open label study, anakinra treatment for 12 weeks significantly decreased non-specific inflammatory measurements, CRP, and erythrocyte sedimentation rate (ESR), and led to a 61% improvement in entheseal lesions at the lumbar spine and sacroiliac joints detected by magnetic resonance imaging [105]. However, in another open label study, anakinra treatment for 24 weeks only led to limited and temporary improvement of clinical symptoms for a small subset of patients [106].
Overall, the above studies demonstrated that the inflammasome and IL-1β are activated in AS patients; however, to date there is lack of evidence for long-term therapeutic benefit of inflammasome/IL-1 inhibition. Moreover, the effects of targeting inflammasome/ IL-1 on osteoproliferation in AS has not been reported in either animal models or AS patients.
Osteoarthritis
Osteoarthritis is a progressive disease featured by cartilage degeneration, subchondral bone modification, crystal deposition, and osteophyte formation [107]. Inflammatory cytokines and catabolic enzymes such as matrix metalloproteinases are associated with synovial inflammation and cartilage degeneration [107].
Among the current OA rodent models, upregulation of NLRP3, caspase-1, GSDMD, IL-1β, and IL-18 had been reported at mRNA and protein level in OA models induced by mechanical stress (meniscectomy) [81, 82] or enzymatic degradation (intraarticular injection of papain) [108]. However, NLRP3 blockade and gene deficiency have shown inconsistent results in OA models. For example, treatment with the NLRP3 inhibitor CY-09, decreased Nlrp3 and Gsdmd expression, and attenuated cartilage damage in the meniscectomy induced OA [81•]. However, Il1b−/− or Nlrp3−/− mice were not protected from cartilage degeneration and osteophyte formation after meniscectomy [82]. Micro crystal deposition is commonly found in the synovium. In a mouse model of OA (induced by intraarticular injection of basic calcium phosphates, the primary crystal found in OA cartilage), OA phenotypes were not prevented in Il1a−/−, Il1b−/−, Pycard−/−, or Nlrp3−/−mice [83]. Intraarticular injection of IL-1β suppressed proteoglycan synthesis and enhanced proteoglycan breakdown in a mouse OA model suggesting IL-1β can induce cartilage degeneration [109].
While OA patient synovial tissue and cartilage biopsies display enhanced IL-1β, IL-18, and caspase-1 expression [110, 111], inflammasome and IL-1 therapies for prevention of cartilage damage or osteophyte formation has not been investigated. The effect of IL-1 blockade (anakinra or AMG108, a humanized monoclonal antibody to IL1R1) on pain relief has been investigated but with disappointing results [112, 113].
Bone Loss Associated Diseases
Inflammatory Arthritis
The term inflammatory arthritis is used to describe a group of autoinflammatory disorders, one of the most recognized is rheumatoid arthritis (RA). RA is an autoimmune inflammatory disease characterized by synovial inflammation, cartilage, and bone erosion which leads to progressive joint deformation [114]. Various pathways have already been identified in promoting RA pathogenesis [115•].
Preclinical models in small animals have been vital in establishing pathological roles for the inflammasome, IL-1β, and IL-18 in RA. Targeting IL1R signaling by administrating anti-IL1α/β or IL1RA ameliorates arthritis in the collagen-induced arthritis (CIA) mouse model [72]. Conversely, unopposed IL-1 signaling, due to the deletion of the Il1ra gene, led to development of spontaneous arthritis [63]. Spontaneous arthritis induced by myeloid-specific deletion of the RA susceptibility gene Tnfaip3 requires inflammasome signaling, as a deficiency in either Nlrp3, casp1/11, or Il1r1 genes in this model rescued arthritic phenotypes [73]. Treatment of CIA mice with the NLRP3 inhibitor MCC950 also attenuated synovial inflammation and bone erosion [74•]. IL-18 expression is also significantly upregulated in the CIA mouse model [75]. Targeting IL-18 via a neutralizing antibody, IL-18 binding protein (IL18BP) or using Il18−/− mice all significantly decreased the severity of arthritis and cartilage damage [75,76,77]. Conversely, intraarticular overexpression of IL-18 leads to joint inflammation and cartilage damage [78].
NLRP3-independent pathways also participate in RA pathogenesis. IL-1β blockade significantly reduced arthritis severity in an antigen-induced arthritis (AIA) mouse model, while Nlrp3 or Nlrc4 gene deletion did not affect AIA [79]. Deletion of the Pycard gene (encoding ASC) protected CIA mice from developing arthritis and bone erosion showing the importance of canonical inflammasomes; however, Nlrp3 and Casp1 null mice developed comparable disease to WT mice [80]. Overall, these studies suggest both NLRP3-dependent and independent pathways (via other canonical inflammasomes) could potentially contribute to IL-18 and IL-1β pathologies in preclinical RA models.
Recent studies have documented an active inflammasome in RA patients. Total leukocytes from RA patients in the acute phase of disease expressed higher levels of ASC, active caspase 1 and NLRP3 compared to healthy controls, and in vitro stimulation of total leukocytes resulted in enhanced IL-1β secretion [116]. IL-18, NLRP3 expression, and caspase-1 activity were also upregulated in RA synovial tissue compared to osteoarthritis (OA) patients [74, 117]. In vitro stimulation of RA synovial fibroblasts with IL-18 increased the production of the chemokines IL-8/CXCL8, epithelial-neutrophil activating protein/CXCL5, growth-regulated oncogene α (GROα)/CXCL1, and the CC chemokine; CCL2 [118] as well as the angiogenic factors stromal cell-derived factor 1α (SDF-1α/CXCL12) and vascular endothelial growth factor A (VEGF-A) [119].
Anakinra was approved for patients with moderate-severe RA who are unresponsive to initial disease-modifying antirheumatic drugs in 2009–2010. Anakinra improved clinical arthritic symptoms and ameliorated radiographic progression [120]; however, anakinra was less effective than the TNF-α antagonist etanercept, with a higher incidence of adverse events [121, 122] and FDA approval was removed. Nevertheless, anti-IL-1 therapy was recommended in RA patients with type 2 diabetes in the TRACK (Treatment of Rheumatoid Arthritis and Comorbidities with Kineret) study as anakinra as it was as efficacious as TNF inhibitors, and enabled a decrease in antidiabetic and glucocorticoid therapies which was not observed in patients treated with TNF inhibitors [123].
Osteoporosis
Osteoporosis is characterized by low bone mass and deterioration of bone architecture, often associated with aging, reduced female hormones due to menopause, and metabolic diseases which disturbs bone remodeling [124].
IL-1β and IL-18 contribute to pathological bone loss in osteoporotic mouse and rat models. Ovariectomized (Ovx) mice mimic postmenopausal-induced bone loss, which can be arrested via IL-1Ra [125, 126] and IL-18BP [84•] administration. Nlrp3 deficiency also significantly slows down femoral bone loss [87]. Bone loss in mouse models of ovariectomy, continuous parathyroid hormone (PTH) infusion, or RANKL-induced osteopenia was also significantly improved in Nlrp3−/− mice [86•].
Studies have demonstrated an association between IL-1β, IL-18, and postmenopausal bone loss in humans. In vitro stimulation of monocytes isolated from healthy premenopausal, untreated healthy postmenopausal, estrogen-treated postmenopausal, and hormone-treated postmenopausal women with osteoporosis showed that menopause significantly increased the capacity of monocytes to produce IL-1β, which was reversed by ovarian hormones [127]. In vitro stimulation of whole blood cells harvested from postmenopausal osteoporosis patients produced higher levels of IL-1β, IL-6, TNF-α, IFN-γ, and GM-CSF compared to healthy controls, and IL-1β levels were negatively correlated with lumbar spine (L2-4) bone mineral density in patients [127, 128]. Moreover, reduction of IL-18BP in PBMCs isolated from osteoporotic patients indicated increased IL-18 signaling in osteoporosis [84•]. Only one clinical study has evaluated IL-1 therapy for osteoporotic patients. In this study, menopausal women were treated with anakinra for 3 weeks without hormone treatment. Serum bone resorption markers were significantly increased in saline-treated patients while anakinra moderately dampened this trend but this was not statistically significant [129]. Therefore, despite IL-1 and IL-18 playing pathological roles in osteoporosis, targeting osteoporosis with IL-1-blocking monotherapy may be insufficient to prevent osteoporosis.
Osteolysis
Joint replacement surgery is an effective way to restore mobility and relieve pain resulting from arthritis, fractures, and avascular necrosis. However, periprosthetic osteolysis, which involves focal bone erosion around implants, is the leading cause of joint replacement failure [130•].
Studies show an increased frequency of IL-1α and IL-1β expressing cells in the pseudo-synovial membrane of the prostheses compared to non-arthritic samples [131]. Serum IL-1β is also associated with early aseptic loosening of hip prosthesis [132]. Orthopedic wear debris has been suggested to initiate inflammation and subsequent osteolysis. In vitro studies demonstrated that metal wear particle debris increase the secretion of IL-1β and IL-18 in mouse bone marrow-derived macrophages [88, 89], which was reduced by inhibiting or knocking-down NLRP3 and caspase-1 [133]. Poly(methyl methacrylate) (PPMA) debris also enhance osteoclast differentiation in vitro [134], and bone marrow derived osteoclast precursors from Nlrp3−/− and Il1r1−/− mice showed less differentiation after exposure to bone particles [86, 90]. Moreover, implantation of PPMA, bone cement, or metal debris in calvaria induced significant bone loss, and Casp1 deficiency or anti-IL-1β treatment significantly ameliorated particles-induced osteolysis [88, 89]. These lines of evidence suggest that inflammasome signaling plays an important role in periprosthetic osteolysis.
Periodontitis
Periodontitis is a disease characterized by inflammation of the tissues surrounding teeth, and is frequently associated with poor oral hygiene. Mouse models of periodontitis were utilized to investigate the role of the inflammasome in periodontal pathology. In a model of tooth ligature-induced periodontitis, Nlrp3−/− mice or mice treated with MCC950 had reduced alveolar bone loss measured by μCT compared to control mice and reduced in vitro osteoclast differentiation [90]. As periodontal diseases are more prevalent in older individuals, one study illustrated that serum IL-1β levels increased with age and were directly proportional to the extent of periodontal destruction. Aged Nlrp3−/− mice developed less alveolar bone loss compared to wild-type mice and continuous administration of MCC950 attenuated the severity of the bone loss, suggesting that NLRP3 inflammasome contributes to periodontitis progression with aging [91•].
Imbalances of the oral microbiome often lead to oral dysbiosis [135], which is a contributing factor to periodontitis. Nlrp3−/− mice challenged with live Porphyromonas gingivalis (P. gingivalis), a Gram-negative oral commensal bacterium, had significantly reduced bone loss after 18 weeks, compared to wild-type mice. Additionally, wild-type mice challenged with P. gingivalis had elevated expression of Il1b and Il18 mRNA in the periodontium [92], which was also noted by others [136]. Bruton’s tyrosine kinase (BTK) is already known to regulate osteoclast maturation and fusion [137, 138], and was shown to also modulate the NLRP3 inflammasome, through interaction with ASC and NLRP3 [139]. Administration of Acalabrutinib a BTK inhibitor, has recently been shown to reduce alveolar bone loss in a P. gingivalis LPS-induced periodontitis mouse model [140]. Whether BTK blockade also impacts inflammasome-driven bone loss remains to be investigated.
In humans, single-nucleotide polymorphisms of the IL1B gene increase susceptibility to periodontitis [141], and gingival tissues from periodontitis patients show elevated mRNA expression of NLRP3 and IL1B genes [142]. MCC950 treatment of human periodontal cells in vitro suppressed LPS-dependent cell death and IL-18 and IL-1β secretion [143]. MCC950 also significantly reduced expression of NLRP3 and Caspase-1 in cell lysates from human periodontal ligament cells [143]. A recent clinical study reported that patients with periodontitis show elevated serum and saliva NLRP3 concentrations [144], and elevated NLRP3, ASC, and IL-1β concentrations in saliva are proposed biomarkers for periodontal clinical status [145]. These accumulating lines of evidence suggest the inflammasome play important roles in periodontitis.
Conclusions
Overall, mounting evidence links the inflammasome/IL1 family activity to bone pathologies. To date, these pathologies are treated by blocking IL-1/IL-1R. While IL-1-blocking therapies can alleviate disease severity, chronic IL-1 blockade can also lead to an increased risk of infections and sepsis, which is why selective targeting of inflammasomes is an attractive future therapeutic strategy. Therapeutic challenges may arise due to redundancies in proteolytic cleavage pathways as well as redundancy of IL-1 isoform signaling. However, there are promising developments in inflammasome therapeutics [146], and new inflammasome/IL-1 based therapies are already in clinical trials for CAPS, RA, and OA (Table 3). There is also ongoing development of new inflammasome small molecule inhibitors, as well as targeting post transcriptional and translational modifications of inflammasomes, all of which will allow for novel therapeutic avenues for managing inflammasome-driven diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27. https://doi.org/10.1111/imr.12621.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. https://doi.org/10.1016/s1097-2765(02)00599-3.
Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217(1):e20190314. https://doi.org/10.1084/jem.20190314.
Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018. https://doi.org/10.1084/jem.20172222.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–98. https://doi.org/10.1038/cr.2015.139.
Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89. https://doi.org/10.1038/s41577-019-0165-0.
Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. 2021;593(7860):607–11. https://doi.org/10.1038/s41586-021-03478-3.
Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Asp Med. 2020;76:100924. https://doi.org/10.1016/j.mam.2020.100924.
Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol. 2016;17(8):906–13. https://doi.org/10.1038/ni.3503.
Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284(29):19420–6. https://doi.org/10.1074/jbc.M901744200.
Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev. 2018;281(1):197–232. https://doi.org/10.1111/imr.12606.
Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev. 2018;281(1):138–53. https://doi.org/10.1111/imr.12616.
Su Z, Tao X. Current understanding of IL-37 in human health and disease. Front Immunol. 2021;12:696605. https://doi.org/10.3389/fimmu.2021.696605.
Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122(12):1722–40. https://doi.org/10.1161/circresaha.118.311362.
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20(1):54–61. https://doi.org/10.1038/nm.3423.
Shao BZ, Cao Q, Liu C. Targeting NLRP3 inflammasome in the treatment of CNS diseases. Front Mol Neurosci. 2018;11:320. https://doi.org/10.3389/fnmol.2018.00320.
Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. 2018;17(1):158. https://doi.org/10.1186/s12943-018-0900-3.
McCall SH, Sahraei M, Young AB, Worley CS, Duncan JA, Ting JP, Marriott I. Osteoblasts express NLRP3, a nucleotide-binding domain and leucine-rich repeat region containing receptor implicated in bacterially induced cell death. J Bone Miner Res. 2008;23(1):30–40. https://doi.org/10.1359/jbmr.071002.
Detzen L, Cheat B, Besbes A, Hassan B, Marchi V, Baroukh B, Lesieur J, Sadoine J, Torrens C, Rochefort G, Bouchet J, Gosset M. NLRP3 is involved in long bone edification and the maturation of osteogenic cells. J Cell Physiol. 2021;236(6):4455–69. https://doi.org/10.1002/jcp.30162.
Alippe Y, Kress D, Ricci B, Sun K, Yang T, Wang C, Xiao J, Abu-Amer Y, Mbalaviele G. Actions of the NLRP3 and NLRC4 inflammasomes overlap in bone resorption. Faseb J. 2021;35(9):e21837. https://doi.org/10.1096/fj.202100767RR.
Akatsu T, Takahashi N, Udagawa N, Imamura K, Yamaguchi A, Sato K, Nagata N, Suda T. Role of prostaglandins in interleukin-1-induced bone resorption in mice in vitro. J Bone Miner Res. 1991;6(2):183–9. https://doi.org/10.1002/jbmr.5650060212.
Lee YM, Fujikado N, Manaka H, Yasuda H, Iwakura Y. IL-1 plays an important role in the bone metabolism under physiological conditions. Int Immunol. 2010;22(10):805–16. https://doi.org/10.1093/intimm/dxq431.
Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res. 1999;247(1):84–93. https://doi.org/10.1006/excr.1998.4320.
Jimi E, Shuto T, Koga T. Macrophage colony-stimulating factor and interleukin-1 alpha maintain the survival of osteoclast-like cells. Endocrinology. 1995;136(2):808–11. https://doi.org/10.1210/endo.136.2.7835314.
Jimi E, Nakamura I, Ikebe T, Akiyama S, Takahashi N, Suda T. Activation of NF-kappaB is involved in the survival of osteoclasts promoted by interleukin-1. J Biol Chem. 1998;273(15):8799–805. https://doi.org/10.1074/jbc.273.15.8799.
Morony S, Capparelli C, Lee R, Shimamoto G, Boone T, Lacey DL, Dunstan CR. A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resorption induced by IL-1beta, TNF-alpha, PTH, PTHrP, and 1, 25(OH)2D3. J Bone Miner Res. 1999;14(9):1478–85. https://doi.org/10.1359/jbmr.1999.14.9.1478.
Udagawa N, Horwood NJ, Elliott J, Mackay A, Owens J, Okamura H, Kurimoto M, Chambers TJ, Martin TJ, Gillespie MT. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. J Exp Med. 1997;185(6):1005–12. https://doi.org/10.1084/jem.185.6.1005.
Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162(2):1096–100.
Makiishi-Shimobayashi C, Tsujimura T, Iwasaki T, Yamada N, Sugihara A, Okamura H, Hayashi SI, Terada N. Interleukin-18 up-regulates osteoprotegerin expression in stromal/osteoblastic cells. Biochem Biophys Res Commun. 2001;281(2):361–6. https://doi.org/10.1006/bbrc.2001.4380.
Simsa-Maziel S, Zaretsky J, Reich A, Koren Y, Shahar R, Monsonego-Ornan E. IL-1RI participates in normal growth plate development and bone modeling. Am J Physiol Endocrinol Metab. 2013;305(1):E15–21. https://doi.org/10.1152/ajpendo.00335.2012.
Vargas SJ, Naprta A, Glaccum M, Lee SK, Kalinowski J, Lorenzo JA. Interleukin-6 expression and histomorphometry of bones from mice deficient in receptors for interleukin-1 or tumor necrosis factor. J Bone Miner Res. 1996;11(11):1736–44. https://doi.org/10.1002/jbmr.5650111117.
Kawase Y, Hoshino T, Yokota K, Kuzuhara A, Nakamura M, Maeda Y, Nishiwaki E, Zenmyo M, Hiraoka K, Aizawa H, Yoshino K. Bone malformations in interleukin-18 transgenic mice. J Bone Miner Res. 2003;18(6):975–83. https://doi.org/10.1359/jbmr.2003.18.6.975.
Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, Rosenberg HF, Austin F, Remmers EF, Balow JE, Rosenzweig S, Komarow H, Shoham NG, Wood G, Jones J, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–8. https://doi.org/10.1002/art.10688.
Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, Basile GS. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Gen. 2002;71(1):198–203. https://doi.org/10.1086/341357.
Hill SC, Namde M, Dwyer A, Poznanski A, Canna S, Goldbach-Mansky R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA). Pediatr Radiol. 2007;37(2):145–52. https://doi.org/10.1007/s00247-006-0358-0.
Houx L, Hachulla E, Kone-Paut I, Quartier P, Touitou I, Guennoc X, et al. Musculoskeletal symptoms in patients with cryopyrin-associated periodic syndromes: a large database study. Arthritis Rheumatol (Hoboken, NJ). 2015;67(11):3027–36. https://doi.org/10.1002/art.39292.
Welzel T, Kuemmerle-Deschner JB. Diagnosis and management of the Cryopyrin-associated periodic syndromes (CAPS): what do we know today? J Clin Med. 2021;10(1):128. https://doi.org/10.3390/jcm10010128.
Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108(4):615–20. https://doi.org/10.1067/mai.2001.118790.
Kuemmerle-Deschner JB. CAPS—pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol. 2015;37(4):377–85. https://doi.org/10.1007/s00281-015-0491-7.
Bonar SL, Brydges SD, Mueller JL, McGeough MD, Pena C, Chen D, et al. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PLoS One. 2012;7(4):e35979. https://doi.org/10.1371/journal.pone.0035979.
Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30(6):875–87. https://doi.org/10.1016/j.immuni.2009.05.005.
Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30(6):860–74. https://doi.org/10.1016/j.immuni.2009.04.012.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–55. https://doi.org/10.1038/nm.3806.
Stackowicz J, Gaudenzio N, Serhan N, Conde E, Godon O, Marichal T, et al. Neutrophil-specific gain-of-function mutations in Nlrp3 promote development of cryopyrin-associated periodic syndrome. J Exp Med. 2021;218(10):e20201466. https://doi.org/10.1084/jem.20201466.
Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 2018;16(11):e3000047. https://doi.org/10.1371/journal.pbio.3000047Proof of principel study illustrating that all NOMID-associated inflammatory symptoms were prevented in mice upon ablation of GSDMD, which supports discovery efforts aimed at identifying GSDMD selective inhibitors.
Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, Zalewski C, Kim HJ, Bishop R, Hill S, Paul SM, Kicker P, Phillips Z, Dolan JG, Widemann B, Jayaprakash N, Pucino F, Stone DL, Chapelle D, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012;64(7):2375–86. https://doi.org/10.1002/art.34409.
Gillespie J, Mathews R, McDermott MF. Rilonacept in the management of cryopyrin-associated periodic syndromes (CAPS). J Inflamm Res. 2010;3:1–8. https://doi.org/10.2147/jir.s8109.
Walker UA, Tilson HH, Hawkins PN, Poll TV, Noviello S, Levy J, et al. Long-term safety and effectiveness of canakinumab therapy in patients with cryopyrin-associated periodic syndrome: results from the β-Confident Registry. RMD open. 2021;7(2):e001663. https://doi.org/10.1136/rmdopen-2021-001663.
Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N Engl J Med. 2003;348(25):2583–4. https://doi.org/10.1056/nejm200306193482523.
Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–85. https://doi.org/10.1016/s0140-6736(04)17401-1.
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92. https://doi.org/10.1056/NEJMoa055137.
Lepore L, Paloni G, Caorsi R, Alessio M, Rigante D, Ruperto N, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J Pediatr. 2010;157(2):310–5.e1. https://doi.org/10.1016/j.jpeds.2010.02.040.
Miyamae T, Inaba Y, Nishimura G, Kikuchi M, Kishi T, Hara R, Kaneko U, Shinoki T, Imagawa T, Yokota S. Effect of anakinra on arthropathy in CINCA/NOMID syndrome. Pediatr Rheumatol Online J. 2010;8:9. https://doi.org/10.1186/1546-0096-8-9.
Timdahl K, Goldbach-Mansky R, Leinonen M, Olivecrona H. THU0514 Effect of long-term treatment with Anakinra (Kineret®) on bone mineral density in patients with severe cryopyrin-associated periodic syndromes. J Ann Rheum Dis. 2015;74(Suppl 2):386. https://doi.org/10.1136/annrheumdis-2015-eular.2367.
Kim YH, Kim BJ, Han J, Choi BY, Lee S. Long-term efficacy of Anakinra in Cryopyrin-associated periodic syndrome: focus on destructive arthropathy. J Clin Immunol. 2021;41:1936–9. https://doi.org/10.1007/s10875-021-01099-z.
Corcoran IH-B SE, Halai R, Domingo-Fernandez R, O'Leary D, Banahan K, Jerala R, Conlon N, Jung T, O'Neill LAJ, Cooper MA, Irvine AD. The NLRP3 inhibitor MCC950 inhibits IL-1β production in PBMC from 19 patients with Cryopyrin-associated periodic syndrome and in 2 patients with Schnitzler’s syndrome. Wellcome Open Res. 2020;5:247. https://doi.org/10.12688/wellcomeopenres.16107.1Provides evidence that the orally available small molecule NLRP3 inhibitor MCC950 blocked constitutive NLRP3 activation in PBMCs from CAPS patients. Highlighting the potential of this small molecule for pathologies driven by NLRP3.
Ren GM, Li J, Zhang XC, Wang Y, Xiao Y, Zhang XY, et al. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci Immunol. 2021;6(58):eabe2933. https://doi.org/10.1126/sciimmunol.abe2933Provides evidence that posttranslational modification of NLRP3 can be targeted via thiolutin treatment, highlighting its potential as a new therapeutic avenue for NLRP3 driven pathologies.
Vande Walle L, Stowe IB, Sacha P, Lee BL, Demon D, Fossoul A, et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019;17(9):e3000354. https://doi.org/10.1371/journal.pbio.3000354.
Macejová Z, Vargová V, Matejka M, Szekanecz Z. Successful treatment of CINCA/NOMID syndrome with interleukin-1 blockade. Israel Med Assoc J IMAJ. 2015;17(6):389–91.
Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, Hessner MJ, Verbsky J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–44. https://doi.org/10.1056/NEJMoa0809568.
Hannum CH, Wilcox CJ, Arend WP, Joslin FG, Dripps DJ, Heimdal PL, Armes LG, Sommer A, Eisenberg SP, Thompson RC. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343(6256):336–40. https://doi.org/10.1038/343336a0.
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgård U, Cowen EW, Pham TH, Booty M, Estes JD, Sandler NG, Plass N, Stone DL, Turner ML, Hill S, Butman JA, Schneider R, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360(23):2426–37. https://doi.org/10.1056/NEJMoa0807865.
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191(2):313–20. https://doi.org/10.1084/jem.191.2.313.
Hirsch E, Irikura VM, Paul SM, Hirsh D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc Natl Acad Sci U S A. 1996;93(20):11008–13. https://doi.org/10.1073/pnas.93.20.11008.
Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191(2):303–12. https://doi.org/10.1084/jem.191.2.303.
Schnellbacher C, Ciocca G, Menendez R, Aksentijevich I, Goldbach-Mansky R, Duarte AM, Rivas-Chacon R. Deficiency of interleukin-1 receptor antagonist responsive to anakinra. Pediatr Dermatol. 2013;30(6):758–60. https://doi.org/10.1111/j.1525-1470.2012.01725.x.
Tseng HW, Kulina I, Girard D, Gueguen J, Vaquette C, Salga M, et al. Interleukin-1 is overexpressed in injured muscles following spinal cord injury and promotes neurogenic heterotopic ossification. J Bone Miner Res. 2022. https://doi.org/10.1002/jbmr.4482Provides evidence of IL-1 driven neurogenic heterotopic ossification in both human and mouse.
Mahy PR, Urist MR. Experimental heterotopic bone formation induced by bone morphogenetic protein and recombinant human interleukin-1B. Clin Orthop Relat Res. 1988;237:236–44.
Yang X, Qu C, Jia J, Zhan Y. NLRP3 inflammasome inhibitor glyburide expedites diabetic-induced impaired fracture healing. Immunobiology. 2019;224(6):786–91. https://doi.org/10.1016/j.imbio.2019.08.008.
Li H, Zhong X, Chen Z, Li W. Suppression of NLRP3 inflammasome improves alveolar bone defect healing in diabetic rats. J Orthop Surg Res. 2019;14(1):167. https://doi.org/10.1186/s13018-019-1215-9.
Lange J, Sapozhnikova A, Lu C, Hu D, Li X, Miclau T 3rd, et al. Action of IL-1beta during fracture healing. J Orthop Res. 2010;28(6):778–84. https://doi.org/10.1002/jor.21061.
Joosten LA, Helsen MM, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum. 1996;39(5):797–809. https://doi.org/10.1002/art.1780390513.
Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512(7512):69–73. https://doi.org/10.1038/nature13322.
Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194(2):231–43. https://doi.org/10.1111/cei.13167This study confirmed MCC950 treatment ameliorated arthritis in the CIA mouse model suggesting targeting the NLRP3 inflammasome with small molecule inhibitors as future therapeutic strategy for RA.
Plater-Zyberk C, Joosten LA, Helsen MM, Sattonnet-Roche P, Siegfried C, Alouani S, et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest. 2001;108(12):1825–32. https://doi.org/10.1172/jci12097.
Wei XQ, Leung BP, Arthur HM, McInnes IB, Liew FY. Reduced incidence and severity of collagen-induced arthritis in mice lacking IL-18. J Immunol. 2001;166(1):517–21. https://doi.org/10.4049/jimmunol.166.1.517.
Banda NK, Vondracek A, Kraus D, Dinarello CA, Kim S-H, Bendele A, et al. Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J Immunol. 2003;170(4):2100–5. https://doi.org/10.4049/jimmunol.170.4.2100.
Joosten LA, Smeets RL, Koenders MI, van den Bersselaar LA, Helsen MM, Oppers-Walgreen B, et al. Interleukin-18 promotes joint inflammation and induces interleukin-1-driven cartilage destruction. Am J Pathol. 2004;165(3):959–67. https://doi.org/10.1016/s0002-9440(10)63357-3.
Kolly L, Karababa M, Joosten LA, Narayan S, Salvi R, Pétrilli V, et al. Inflammatory role of ASC in antigen-induced arthritis is independent of caspase-1, NALP-3, and IPAF. J Immunol. 2009;183(6):4003–12. https://doi.org/10.4049/jimmunol.0802173.
Ippagunta SK, Brand DD, Luo J, Boyd KL, Calabrese C, Stienstra R, van de Veerdonk FL, Netea MG, Joosten LAB, Lamkanfi M, Kanneganti TD. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J Biol Chem. 2010;285(16):12454–62. https://doi.org/10.1074/jbc.M109.093252.
Zhang Y, Lin Z, Chen D, He Y. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. 2021;553:119–25. https://doi.org/10.1016/j.bbrc.2021.03.055This article shows targeting the NLRP3 inflammasome with the small molecule inhibitor Cy-09, attenuates inflammation and cartilage damage in the meniscectomy induced OA mouse model.
Nasi S, Ea HK, So A, Busso N. Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and -1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front Pharmacol. 2017;8:282. https://doi.org/10.3389/fphar.2017.00282.
Ea HK, Chobaz V, Nguyen C, Nasi S, van Lent P, Daudon M, Dessombz A, Bazin D, McCarthy G, Jolles-Haeberli B, Ives A, van Linthoudt D, So A, Lioté F, Busso N. Pathogenic role of basic calcium phosphate crystals in destructive arthropathies. PLoS One. 2013;8(2):e57352. https://doi.org/10.1371/journal.pone.0057352.
Mansoori MN, Shukla P, Kakaji M, Tyagi AM, Srivastava K, Shukla M, et al. IL-18BP is decreased in osteoporotic women: Prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci Rep. 2016;6:33680. https://doi.org/10.1038/srep33680This study provides evidence of the association between IL-18/IL-18BP and BMD in post menopausal women. Both in vitro and in vivo (ovarieoctomy mouse model) studies suggests IL18BP as a potential treatment for postmenopausal osteoporosis.
Kitazawa R, Kimble RB, Vannice JL, Kung VT, Pacifici R. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J Clin Invest. 1994;94(6):2397–406. https://doi.org/10.1172/jci117606.
Alippe Y, Wang C, Ricci B, Xiao J, Qu C, Zou W, et al. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci Rep. 2017;7(1):6630. https://doi.org/10.1038/s41598-017-07014-0This article confirmed that signals originating from bone matrix activate NLRP3 in osteoclasts. Deficiency in the Nlrp3 gene ameliorated various osteopenic mouse models providing a mechanism that amplifies bone resorption in diseases with pathologic bone loss.
Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–32. https://doi.org/10.1016/j.cmet.2013.09.010.
Burton L, Paget D, Binder NB, Bohnert K, Nestor BJ, Sculco TP, Santambrogio L, Ross FP, Goldring SR, Purdue PE. Orthopedic wear debris mediated inflammatory osteolysis is mediated in part by NALP3 inflammasome activation. J Orthop Res. 2013;31(1):73–80. https://doi.org/10.1002/jor.22190.
Eger M, Hiram-Bab S, Liron T, Sterer N, Carmi Y, Kohavi D, et al. Mechanism and prevention of titanium particle-induced inflammation and osteolysis. Front Immunol. 2018;9:2963. https://doi.org/10.3389/fimmu.2018.02963.
Chen Y, Yang Q, Lv C, Chen Y, Zhao W, Li W, Chen H, Wang H, Sun W, Yuan H. NLRP3 regulates alveolar bone loss in ligature-induced periodontitis by promoting osteoclastic differentiation. Cell Prolif. 2021;54(2):e12973. https://doi.org/10.1111/cpr.12973.
Zang Y, Song JH, Oh SH, Kim JW, Lee MN, Piao X, et al. Targeting NLRP3 Inflammasome reduces age-related experimental alveolar bone loss. J Dent Res. 2020;99(11):1287–95. https://doi.org/10.1177/0022034520933533Provides evidence that NLRP3 activation contributes to periodontitis progression associated with aging, and MCC950 treatment or NLRP3 deficiency reduces age-dependent alveolar bone loss.
Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Regulation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated periodontal disease. Inflamm Res. 2017;66(1):59–65. https://doi.org/10.1007/s00011-016-0992-4.
Vanden Bossche L, Vanderstraeten G. Heterotopic ossification: a review. J Rehabil Med. 2005;37(3):129–36.
Ohlmeier M, Suero EM, Aach M, Meindl R, Schildhauer TA, Citak M. Muscle localization of heterotopic ossification following spinal cord injury. Spine J. 2017;17(10):1519–22. https://doi.org/10.1016/j.spinee.2017.04.021.
Genêt F, Minooee K, Jourdan C, Ruet A, Denormandie P, Schnitzler A. Troublesome heterotopic ossification and stroke: features and risk factors. A case control study. Brain Injury. 2018;0(0):1–6. https://doi.org/10.3109/02699052.2015.1005133.
Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267(5206):2000–3. https://doi.org/10.1126/science.7535475.
Sung Hsieh HH, Chung MT, Allen RM, Ranganathan K, Habbouche J, Cholok D, Butts J, Kaura A, Tiruvannamalai-Annamalai R, Breuler C, Priest C, Loder SJ, Li J, Li S, Stegemann J, Kunkel SL, Levi B. Evaluation of salivary cytokines for diagnosis of both trauma-induced and genetic heterotopic ossification. Front Endocrinol. 2017;8:74. https://doi.org/10.3389/fendo.2017.00074.
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525–7. https://doi.org/10.1038/ng1783.
Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, Kaplan FS, Shore EM. Depletion of mast cells and macrophages impairs heterotopic ossification in an Acvr1(R206H) mouse model of fibrodysplasia ossificans progressiva. J Bone Miner Res. 2018;33(2):269–82. https://doi.org/10.1002/jbmr.3304.
Haviv R, Moshe V, De Benedetti F, Prencipe G, Rabinowicz N, Uziel Y. Is fibrodysplasia ossificans progressiva an interleukin-1 driven auto-inflammatory syndrome? Pediatr Rheumatol Online J. 2019;17(1):84. https://doi.org/10.1186/s12969-019-0386-6.
Wang X, Friis TE, Masci PP, Crawford RW, Liao W, Xiao Y. Alteration of blood clot structures by interleukin-1 beta in association with bone defects healing. Sci Rep. 2016;6(1):35645. https://doi.org/10.1038/srep35645.
Olmedo ML, Landry PS, Sadasivan KK, Albright JA, Meek WD, Routh R, Marino AA. Regulation of osteoblast levels during bone healing. J Orthop Trauma. 1999;13(5):356–62. https://doi.org/10.1097/00005131-199906000-00006.
Tan S, Wang R, Ward MM. Syndesmophyte growth in ankylosing spondylitis. Curr Opin Rheumatol. 2015;27(4):326–32. https://doi.org/10.1097/BOR.0000000000000179.
Guggino G, Mauro D, Rizzo A, Alessandro R, Raimondo S, Bergot A-S, et al. Inflammasome activation in ankylosing spondylitis is associated with gut dysbiosis. Arthritis Rheumatol. 2021;73(7):1189–99. https://doi.org/10.1002/art.41644.
Tan AL, Marzo-Ortega H, O'Connor P, Fraser A, Emery P, McGonagle D. Efficacy of anakinra in active ankylosing spondylitis: a clinical and magnetic resonance imaging study. Ann Rheum Dis. 2004;63(9):1041–5. https://doi.org/10.1136/ard.2004.020800.
Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis. 2005;64(2):296–8. https://doi.org/10.1136/ard.2004.023176.
Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13(5):302–11. https://doi.org/10.1038/nrrheum.2017.50.
Cheng F, Yan FF, Liu YP, Cong Y, Sun KF, He XM. Dexmedetomidine inhibits the NF-κB pathway and NLRP3 inflammasome to attenuate papain-induced osteoarthritis in rats. Pharm Biol. 2019;57(1):649–59. https://doi.org/10.1080/13880209.2019.1651874.
van Beuningen HM, Arntz OJ, van den Berg WB. In vivo effects of interleukin-1 on articular cartilage. Prolongation of proteoglycan metabolic disturbances in old mice. Arthritis Rheum. 1991;34(5):606–15. https://doi.org/10.1002/art.1780340513.
Saha N, Moldovan F, Tardif G, Pelletier JP, Cloutier JM, Martel-Pelletier J. Interleukin-1beta-converting enzyme/caspase-1 in human osteoarthritic tissues: localization and role in the maturation of interleukin-1beta and interleukin-18. Arthritis Rheum. 1999;42(8):1577–87. https://doi.org/10.1002/1529-0131(199908)42:8<1577::Aid-anr3>3.0.Co;2-z.
Koh SM, Chan CK, Teo SH, Singh S, Merican A, Ng WM, Abbas A, Kamarul T. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. The Knee. 2020;27(1):26–35. https://doi.org/10.1016/j.knee.2019.10.028.
Chevalier X, Goupille P, Beaulieu AD, Burch FX, Bensen WG, Conrozier T, Loeuille D, Kivitz AJ, Silver D, Appleton BE. Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009;61(3):344–52. https://doi.org/10.1002/art.24096.
Cohen SB, Proudman S, Kivitz AJ, Burch FX, Donohue JP, Burstein D, Sun YN, Banfield C, Vincent MS, Ni L, Zack DJ. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res Ther. 2011;13(4):R125. https://doi.org/10.1186/ar3430.
Auréal M, Machuca-Gayet I, Coury F. Rheumatoid arthritis in the view of osteoimmunology. Biomolecules. 2020;11(1):48. https://doi.org/10.3390/biom11010048.
Makkar R, Behl T, Bungau S, Kumar A, Arora S. Understanding the role of inflammasomes in rheumatoid arthritis. Inflammation. 2020;43(6):2033–47. https://doi.org/10.1007/s10753-020-01301-1. This review highlights the roles of different inflammasomes in RA and their potential function in disease.
Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, Bertsias G, Boumpas DT, Sidiropoulos P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):257. https://doi.org/10.1186/s13075-015-0775-2.
Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, Kennedy K, Carter R, Wei XQ, Xu D, Field M, Foulis A, Liew FY, McInnes IB. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104(10):1393–401. https://doi.org/10.1172/jci7317.
Morel JC, Park CC, Kumar P, Koch AE. Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation. Lab Invest. 2001;81(10):1371–83. https://doi.org/10.1038/labinvest.3780351.
Amin MA, Mansfield PJ, Pakozdi A, Campbell PL, Ahmed S, Martinez RJ, Koch AE. Interleukin-18 induces angiogenic factors in rheumatoid arthritis synovial tissue fibroblasts via distinct signaling pathways. Arthritis Rheum. 2007;56(6):1787–97. https://doi.org/10.1002/art.22705.
Bresnihan B, Cobby M. Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology. 2003;42(suppl_2):ii22–i8. https://doi.org/10.1093/rheumatology/keg329.
Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36(6):1118–25. https://doi.org/10.3899/jrheum.090074.
Ramírez J, Cañete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Exp Opin Drug Saf. 2018;17(7):727–32. https://doi.org/10.1080/14740338.2018.1486819.
Ruscitti P, Berardicurti O, Cipriani P, Giacomelli R. Benefits of anakinra versus TNF inhibitors in rheumatoid arthritis and type 2 diabetes: long-term findings from participants furtherly followed-up in the TRACK study, a multicentre, open-label, randomised, controlled trial. Clin Exp Rheumatol. 2021;39(2):403–6.
Anagnostis P, Gkekas NK, Potoupnis M, Kenanidis E, Tsiridis E, Goulis DG. New therapeutic targets for osteoporosis. Maturitas. 2019;120:1–6. https://doi.org/10.1016/j.maturitas.2018.11.010.
Kimble RB, Vannice JL, Bloedow DC, Thompson RC, Hopfer W, Kung VT, Brownfield C, Pacifici R. Interleukin-1 receptor antagonist decreases bone loss and bone resorption in ovariectomized rats. J Clin Invest. 1994;93(5):1959–67. https://doi.org/10.1172/jci117187.
Baltzer AW, Whalen JD, Wooley P, Latterman C, Truchan LM, Robbins PD, Evans CH. Gene therapy for osteoporosis: evaluation in a murine ovariectomy model. Gene Ther. 2001;8(23):1770–6. https://doi.org/10.1038/sj.gt.3301594.
Pacifici R, Rifas L, McCracken R, Avioli LV. The role of interleukin-1 in postmenopausal bone loss. Exp Gerontol. 1990;25(3-4):309–16. https://doi.org/10.1016/0531-5565(90)90067-c.
Zheng SX, Vrindts Y, Lopez M, De Groote D, Zangerle PF, Collette J, et al. Increase in cytokine production (IL-1β, IL-6, TNF-α but not IFN-γ, GM-CSF or LIF) by stimulated whole blood cells in postmenopausal osteoporosis. Maturitas. 1997;26(1):63–71. https://doi.org/10.1016/S0378-5122(96)01080-8.
Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res. 2007;22(5):724–9. https://doi.org/10.1359/jbmr.070207.
Mbalaviele G, Novack DV, Schett G, Teitelbaum SL. Inflammatory osteolysis: a conspiracy against bone. J Clin Invest. 2017;127(6):2030–9. https://doi.org/10.1172/jci93356A review providing an overview of the types of osteolysis and the role of inflammasomes in osteolysis.
Westacott CI, Taylor G, Atkins R, Elson C. Interleukin 1 alpha and beta production by cells isolated from membranes around aseptically loose total joint replacements. Ann Rheum Dis. 1992;51(5):638–42. https://doi.org/10.1136/ard.51.5.638.
Wang Y, Li G, Ma J, Ji B, Aishajiang A, Yelborati B, Cao L. Effect of interleukin 1 receptor antagonist gene on stable expression bone marrow mesenchymal stem cells and early aseptic loosening of hip prosthesis of mouse. Mol Biotechnol. 2021;63(3):232–9. https://doi.org/10.1007/s12033-020-00297-1.
Jämsen E, Pajarinen J, Kouri VP, Rahikkala A, Goodman SB, Manninen M, Nordström DC, Eklund KK, Nurmi K. Tumor necrosis factor primes and metal particles activate the NLRP3 inflammasome in human primary macrophages. Acta Biomaterialia. 2020;108:347–57. https://doi.org/10.1016/j.actbio.2020.03.017.
Clohisy JC, Frazier E, Hirayama T, Abu-Amer Y. RANKL is an essential cytokine mediator of polymethylmethacrylate particle-induced osteoclastogenesis. J Orthop Res. 2003;21(2):202–12. https://doi.org/10.1016/s0736-0266(02)00133-x.
Radaic A, Kapila YL. The oralome and its dysbiosis: new insights into oral microbiome-host interactions. Comput Struct Biotechnol J. 2021;19:1335–60. https://doi.org/10.1016/j.csbj.2021.02.010.
Zhang Y, Kuang W, Li D, Li Y, Feng Y, Lyu X, Huang GB, Lian JQ, Yang XF, Hu C, Xie Y, Xue S, Tan J. Natural killer-like B cells secreting interleukin-18 induces a proinflammatory response in periodontitis. Front Immunol. 2021;12:641562. https://doi.org/10.3389/fimmu.2021.641562.
Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk regulates RANKL-induced osteoclast maturation. J Biol Chem. 2008;283(17):11526–34. https://doi.org/10.1074/jbc.M708935200.
Shinohara M, Chang BY, Buggy JJ, Nagai Y, Kodama T, Asahara H, Takayanagi H. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone. 2014;60:8–15. https://doi.org/10.1016/j.bone.2013.11.025.
Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360. https://doi.org/10.1038/ncomms8360.
Pokhrel NK, Kim YG, Kim HJ, Kim HJ, Lee JH, Choi SY, Kwon TG, Lee HJ, Kim JY, Lee Y. A novel Bruton's tyrosine kinase inhibitor, acalabrutinib, suppresses osteoclast differentiation and Porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption. J Periodontol. 2019;90(5):546–54. https://doi.org/10.1002/JPER.18-0334.
Offenbacher S, Jiao Y, Kim SJ, Marchesan J, Moss KL, Jing L, Divaris K, Bencharit S, Agler CS, Morelli T, Zhang S, Sun L, Seaman WT, Cowley D, Barros SP, Beck JD, Munz M, Schaefer AS, North KE. GWAS for Interleukin-1beta levels in gingival crevicular fluid identifies IL37 variants in periodontal inflammation. Nat Commun. 2018;9(1):3686. https://doi.org/10.1038/s41467-018-05940-9.
Aral K, Berdeli E, Cooper PR, Milward MR, Kapila Y, Karadede Unal B, et al. Differential expression of inflammasome regulatory transcripts in periodontal disease. J Periodontol. 2020;91(5):606–16. https://doi.org/10.1002/JPER.19-0222.
Peng W, Zhang B, Sun Z, Zhang M, Guo L. Targeting the Nod-like receptor protein 3 Inflammasome with inhibitor MCC950 rescues lipopolysaccharide-induced inhibition of osteogenesis in Human periodontal ligament cells. Arch Oral Biol. 2021;131:105269. https://doi.org/10.1016/j.archoralbio.2021.105269.
Isola G, Polizzi A, Santonocito S, Alibrandi A, Williams RC. Periodontitis activates the NLRP3 inflammasome in serum and saliva. J Periodontol. 2021;93:135–45. https://doi.org/10.1002/JPER.21-0049.
Isaza-Guzman DM, Medina-Piedrahita VM, Gutierrez-Henao C, Tobon-Arroyave SI. Salivary levels of NLRP3 inflammasome-related proteins as potential biomarkers of periodontal clinical status. J Periodontol. 2017;88(12):1329–38. https://doi.org/10.1902/jop.2017.170244.
Caseley EA, Poulter JA, Rodrigues F, McDermott MF. Inflammasome inhibition under physiological and pharmacological conditions. Genes and immunity. 2020;21(4):211–23. https://doi.org/10.1038/s41435-020-0104-x.
Mullard A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat Rev Drug Discov. 2019;18(6):405–7. https://doi.org/10.1038/d41573-019-00086-9.
Current Clinical Trials for OLT1177® Dapansutrile. http://www.olatec.com/patients.html Accessed.
Switching off chronic inflammation. Nature Portfolio.
Acknowledgements
JPL and KS are supported by Research Fellowships 1136130 and 1141131 from the National Health and Medical Research Council of Australia (NHMRC). JPL, KAA, HWT research is supported by NHMRC Ideas Grant 1181053. KAA is also supported by a University of Queensland Early Career Researcher Grant UQECR1834805 and by funds from the Mater Foundation. The caspase-1 null mice (B6N.129S2-Casp1tm1Flv/J) were kindly provided by Prof Richard A. Flavell [96]. MCC950 was kindly provided by Prof. Avril Robertson [43]. The authors also acknowledge the scientific and technical assistance at the Translational Research Institute: The University of Queensland biological resources TRI facility and the preclinical imaging facility, which is supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
HWT, JPL, and KAA declare no conflict of interest. KS is a co-inventor on patent applications for NLRP3 inhibitors which have been licensed to Inflazome Ltd, a company headquartered in Dublin, Ireland. Inflazome is developing drugs that target the NLRP3 inflammasome to address unmet clinical needs in inflammatory disease. K.S. served on the Scientific Advisory Board of Inflazome in 2016–2017, and serves as a consultant to Quench Bio, USA and Novartis, Switzerland.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Osteoimmunology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tseng, HW., Samuel, S.G., Schroder, K. et al. Inflammasomes and the IL-1 Family in Bone Homeostasis and Disease. Curr Osteoporos Rep 20, 170–185 (2022). https://doi.org/10.1007/s11914-022-00729-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-022-00729-8