
Abstract
Purpose of Review
This review summarizes current understanding of generalized arterial calcification of infancy (GACI), emphasizing pathophysiology, clinical presentation, and approaches and controversies in management.
Recent Findings
Identification of causative ENPP1 mutations revealed that GACI arises from deficiencies in inorganic pyrophosphate (leading to calcifications) and adenosine monophosphate (leading to intimal proliferation). Identification of genotypic and phenotypic overlap with pseudoxanthoma elasticum and autosomal recessive hypophosphatemic rickets further advanced understanding of GACI as a complex, multisystemic disease. Clinical data is limited to small, retrospective samples; it is therefore unknown whether commonly used medications, such as bisphosphonates and hypophosphatemia treatment, are therapeutic or potentially harmful. ENPP1-Fc replacement represents a promising approach warranting further study.
Summary
Knowledge gaps in natural history place clinicians at high risk of assigning causality to interventions that are correlated with changes in clinical status. There is thus a critical need for improved natural history studies to develop and test targeted therapies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Generalized arterial calcification of infancy (GACI) is a rare, life-threatening disorder arising from deficiencies in ENPP1 or ABCC6. Patients develop calcifications and intimal proliferation in multiple arteries, often resulting in critical illness and death in utero or early infancy. Recent advances identifying key genetic and pathophysiologic features of GACI have broadened our understanding of its complex, multisystemic phenotype, which may include calcifications and other extravascular features in varying degrees. However, critical gaps in understanding the natural history of GACI have severely limited the development of evidence-based treatment strategies. This review will provide a background on the pathophysiology and clinical presentation in GACI, discuss current approaches and controversies in management, and outline key areas of need for ongoing and future research.
Pathophysiology
Approximately 100 years after GACI was first reported in the literature [1], a genetic cause was identified. Rutsch and colleagues recognized that the tiptoe walking (ttw/ttw) mice, caused by spontaneous inactivating variants in the gene Npps [2], had features similar to those seen in children with GACI, such as periarticular and aortic calcifications, decreased inorganic pyrophosphate (PPi), and impaired ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) activity [3]. A study of 11 children with GACI from unrelated kindreds revealed biallelic loss-of-function variants distributed throughout the coding regions of ENPP1 in 8 individuals [4]; subsequent studies confirmed these findings with an estimated 75% of GACI cases due to ENPP1 mutations [5, 6].
ENPP1 is a transmembrane glycoprotein that hydrolyzes extracellular adenosine triphosphate (ATP) to form adenosine monophosphate (AMP) and inorganic pyrophosphate (PPi). PPi is a potent inhibitor of mineralization, functioning as a physiologic “water-softener” by preventing both the formation and growth of hydroxyapatite crystals [7]. Deficiency of ENPP1, as seen in patients with GACI, results in decreased generation of extracellular PPi leading to ectopic calcification, particularly in the vascular internal elastic lamina, cartilage, and other soft tissues [3, 7]. However, the vascular stenosis observed in babies with GACI is not due solely to the presence of calcium deposits in the vessel wall; there is vascular smooth muscle cell proliferation, further decreasing lumen diameter and compromising blood flow [3]. Recent animal studies suggest that it is the concurrent lack of AMP generation in ENPP1 deficiency that leads to this neointimal proliferation [8••].
In addition to impaired production of extracellular AMP and PPi, many individuals with ENPP1 deficiency who survive beyond infancy develop hypophosphatemia (autosomal recessive hypophosphatemic rickets type 2) due to elevation of fibroblast growth factor 23 (FGF23) [9, 10]. Enpp1−/− knockout mice have reduced osteocyte lacunar size, narrowed canaliculi, and decreased mechanical loading which is associated with increased circulating sclerostin; it has been speculated that this increase in sclerostin may upregulate FGF23 expression by bone cells [11]. However, the definitive mechanism for the increased FGF23 in both animal models and humans is currently unknown.
Several years later, inactivating mutations in the gene encoding ATP-binding cassette subfamily C member 6 (ABCC6) were identified as a second genetic cause of GACI [12]. Further examination of cohorts with ENPP1 and ABCC6 deficiency revealed that both GACI and the rare disorder pseudoxanthoma elasticum (PXE) could be caused by loss-of-function variants in either gene, with an overlapping spectrum and variable phenotype [12, 13]. ABCC6 traverses the cell membrane, presumably transporting nucleotide triphosphates, possibly ATP, into the extracellular space. However, the exact role ABCC6 deficiency plays in the mineralization cascade is currently unknown.
A distinct but related condition is arterial calcification due to deficiency of CD73 (ACDC), an autosomal recessive disorder due to loss-of-function variants in NT5E, the gene encoding CD73. CD73 is an endonucleotidase that converts AMP to adenosine; CD73 deficiency results in progressive adult-onset vascular calcifications and ischemia in the lower extremities and articular calcifications [14]. In vitro studies suggest that this reduction in adenosine leads to increased TNAP activity which reduces PPi and promotes calcification [15].
Clinical Presentation
GACI is a multisystemic disease with a complex phenotype (Table 1). The signature manifestation is vascular calcification, thought to account for the most serious morbidity and mortality. However, disease may affect various other systems, particularly in patients who survive beyond infancy. In addition, increasing recognition of genotypic and phenotypic overlap between GACI and other disorders is further expanding our understanding of the broad clinical spectrum in this challenging disease.
Vascular Calcifications
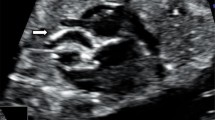
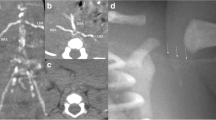
Vascular calcifications are the earliest and most dramatic feature of GACI (Fig. 1a–d). In a comprehensive literature review by Chong et al. involving 161 patients, 48% presented in utero or shortly after birth, termed “early onset,” and 52% presented after birth (median 3 months), termed “late onset” [16]. These groups had both distinct and overlapping clinical features. In early-onset GACI, patients most commonly presented with signs of fetal circulatory compromise, including hydrops fetalis, polyhydramnios, and fetal distress. Patients with late-onset disease were often asymptomatic at birth, but developed progressive signs of cardiovascular disease, including respiratory distress, cyanosis, and feeding difficulties.

Clinical images of vascular calcifications in patients with GACI. a Coronal views of a computed tomography scan shows diffuse calcification involving the descending aorta (red arrow) and multiple branching arteries (yellow asterisks). b Prenatal ultrasound demonstrates calcification and severe narrowing in the distal aorta (red arrow). c Axial views of a computed tomography scan show a calcified and severely narrowed descending aorta (red arrow). d Medium-sized pancreatic artery showing calcification (red arrows) along the internal elastic lamina (hematoxylin and eosin stain)
A wide variety of arteries may be affected by calcifications. In the Chong et al. review, autopsies revealed that early-onset disease most commonly involved the hepatic (81%), aortic (80%), and pulmonary arteries (67%), while in late-onset disease, the coronary (88%), renal (55%), and pulmonary arteries (49%) were most affected [16]. However, these results reflect autopsies performed at numerous centers over an extensive period, and standardized studies with well-defined disease criterion are needed to characterize arterial involvement.
Peripheral arterial calcifications are common and generally present with diminished peripheral pulses [12]; ischemic ulcerations leading to gangrene are a rare but established complication [17, 18]. Persistent pulmonary hypertension with increased pulmonary vascular resistance has been reported in neonates [19, 20]. Intestinal ischemia can result in a variety of gastrointestinal complications, including obstruction, peritonitis, and ulcerative inflammation [21]. Involvement of cerebral arteries is uncommonly reported, but may result in serious neurologic morbidity, including seizures, strokes, and encephalomalacia [12, 22,23,24].
Extravascular Calcifications
Calcifications may involve a variety of extravascular sites, including the myocardium, pancreas, liver, and kidneys, which may potentially contribute to multiorgan dysfunction [12, 25]. Periarticular calcifications appear to have a predilection for the hips, ankles, wrists, and shoulders [16](Fig. 2c, d) and may result in pain and reduced range of motion. Interestingly, there are several reports of calcifications involving the earlobes, making this a potentially useful physical exam finding, although it is not universally present [12, 26, 27].

Clinical images of extravascular features in patients with GACI. a Skin findings consistent with PXE. Note the characteristic yellowish papules occurring in a patchy, reticular pattern (white arrows). b Fundus autofluorescence imaging in an adult patient with ENPP1 deficiency revealing a large area of hypoautofluorescence (red arrowheads) due to atrophy of the retinal pigment epithelium, with spots of hyperautofluorescence surrounding the area of atrophy. c Large area of periarticular calcifications involving the shoulder joint (arrow). d Periarticular calcifications involving the ankle joint (arrow). e Irregularities in the distal femoral metaphyses consistent with hypophosphatemic rickets (red arrowheads). Note the bowing of the bilateral femoral shafts (white arrows), resulting in a genu valgum deformity of the knees
Relationship with Pseudoxanthoma Elasticum
GACI and PXE share intriguing genetic and phenotypic similarities. PXE is a connective tissue disorder associated with progressive mineralization and fragmentation of elastic fibers in the skin, eye, and cardiovascular systems, typically arising due to biallelic mutations in ABCC6 [28]. Characteristic clinical features include retinal angioid streaks (Fig. 2b), arising from disruption of the elastin-rich membrane between the retina and choriocapillaris, and yellowish skin papules (Fig. 2a), arising from calcification of fragmented elastic fibers. Arterial calcification is a well-established feature of PXE that results in a variety of complications, including coronary artery disease, ischemic strokes, and peripheral vascular disease [28].
An analysis of an international GACI registry identified biallelic and monoallelic ABCC6 mutations in 8/92 and 6/92 patients, respectively, all of whom presented with classic symptoms of severe infantile arteriopathy [12]. Typical ocular and cutaneous features of PXE have also been reported in individuals with GACI secondary to ENPP1 deficiency [12, 29]. This genotypic and phenotypic overlap indicates that ABCC6 and ENPP1 mutations likely impact similar physiological processes, suggesting that GACI and PXE are more appropriately conceptualized as components of a clinical spectrum rather than two distinct disorders.
Relationship with Autosomal Recessive Hypophosphatemic Rickets Type 2
GACI also appears to share interesting genotypic and phenotypic similarities with autosomal recessive hypophosphatemic rickets type 2 (ARHR2). This FGF23 excess disorder also arises due to inactivating ENPP1 mutations and typically presents in childhood with renal phosphate wasting leading to rickets, skeletal deformities, muscle weakness, and bone pain [9, 10] (Fig. 2e). Patients with GACI due to ENPP1 deficiency who survive the neonatal period may develop hypophosphatemia later in life, with a clinical presentation consistent with ARHR2 [12, 30]. The relationship between ARHR2 and vascular calcifications is unclear. Although there are no reports of clinically significant cardiovascular compromise in patients with ARHR2, it is possible that patients may have had asymptomatic, undiagnosed calcifications in infancy that resolved after the onset of hypophosphatemia. GACI and ARHR2 therefore also represent components of a clinical spectrum of ENPP1 deficiency. The relationship between hypophosphatemia and ABCC6 mutations is comparatively less well established; there is only one reported case of hypophosphatemic rickets in a patient with GACI with a monoallelic ABCC6 mutation [12].
Hearing Loss
Early-onset hearing loss can be associated with GACI arising from ENPP1 mutations and appears to be multifactorial in origin. One patient developed sensorineural hearing loss at age 4; the authors postulated this may be related to calcification of the arteries supplying the inner ear [9]. Two children developed conductive hearing loss due to stapediovestibular ankylosis [12], and one developed mixed hearing loss, potentially due to effects of calcification on both the inner and middle ear compartments [26].
Interestingly, hearing loss has not yet been reported in patients with GACI caused by ABCC6 mutations and does not appear to be a typical feature of classical PXE.
Dental Disease
Abnormalities in dental development have been reported in patients with GACI related to ENPP1 deficiency, including infraocclusion, retained primary teeth, ankylosis, and slow orthodontic tooth movement [31]. Analyses of primary teeth revealed dramatically expanded cervical cementum, supporting a key role of pyrophosphate in cementogenesis.
Other Manifestations
Clinical descriptions in GACI have been defined entirely by small and retrospective studies, which have limited understanding of its full phenotypic spectrum. Recently, novel bilallelic ENPP1 mutations were identified in 4 infants from 2 consanguineous families presenting with severe multisystemic disease [32]. Infants were born prematurely with a phenotype mimicking congenital infection, including severe thrombocytopenia, hypoglycemia, liver disease, and significant brain abnormalities, resulting in death in the first several months. Arterial calcifications were not initially recognized as a prominent feature in these patients. However, after ENPP1 mutations were revealed by whole exome sequencing, re-analyses of imaging studies noted multiple calcifications in large- and medium-sized arteries, establishing a clinical diagnosis of GACI. As demonstrated in this series, increased use of genomic sequences technologies is likely to uncover additional novel phenotypic associations in patients with GACI.
Genotype-Phenotype Correlations
Genotype-phenotype correlations in GACI have not been clearly established. Evidence suggests that hypophosphatemia and hearing loss are more closely associated with ENPP1 as opposed to ABCC6 mutations (Table 1); however, larger phenotyping studies are needed to confirm these correlations. A retrospective series of 55 infants identified homozygosity in the ENPP1 variant p.Pro305Thr as associated with infantile death in all patients [5]; however, other series have reported markedly divergent clinical courses in siblings with identical mutations [33, 34]. In one infant with GACI, bilallelic ENPP1 mutations and a monoallelic ABCC6 mutation were identified, which theoretically could have contributed to a more severe phenotype [35]. However, this patient’s healthy mother and sibling were heterozygous for ENPP1 and ABCC6 mutations, suggesting this digenic inheritance likely does not cause GACI.
Prognosis
GACI is life-threatening with a historically poor prognosis. A retrospective series reported neonatal death in 30/55 patients, including 6 stillbirths [5]. Deaths were related to cardiovascular compromise, including myocardial infarction, congestive heart failure, persistent pulmonary hypertension, and multiorgan failure. All but one death occurred in the first 6 months, indicating that the mortality rate decreases markedly after infancy. Factors associated with survival in this series included hypophosphatemia and bisphosphonate treatment (discussed in detail in subsequent sections).
Life expectancy for patients who survive the neonatal period has not been established. There are several reports of long-term survivors in their third decade [5, 36], as well as deaths in childhood [37]. Spontaneous resolution of calcifications has been documented in several cases [38, 39]. One teenage patient with a history of infantile disease underwent radial artery biopsy showing resolution of previous calcifications, but intimal thickening resembling fibromuscular dysplasia [40], suggesting GACI may have potential long-term vascular effects. This possibility is supported by additional cases documenting arterial stenoses without calcifications in long-term survivors [12, 41].
Management
Critical knowledge gaps have limited the development of consistent management strategies in GACI. There are no prospective trials, and available retrospective studies were performed across multiple centers without standardized care or systematic data collection. The following sections summarize the most common approaches to management, including rationale, supporting evidence, and areas of controversy. In addition, Table 2 includes a brief summary of the authors’ suggested approach to management and monitoring.
Bisphosphonates
Bisphosphonates have been used in the treatment of GACI since the late 1970s [42]. These medications are structural analogs of pyrophosphate with a high affinity for hydroxyapatite. They are incorporated preferentially into sites of active bone remodeling, where they have the potential to inhibit both calcification and hydroxyapatite breakdown [43]. The rationale for use of bisphosphonates in GACI initially arose after electron diffraction studies demonstrated that calcium deposits consist of hydroxyapatite [42]. In the 1990s, pyrophosphate deficiency was established as part of the pathophysiology of GACI [44], further suggesting a potential role for bisphosphonates.
Bisphosphonates consist of a core P-C-P motif as opposed to the P-O-P structure found in pyrophosphate, with two side chains that determine the anti-resorptive properties that distinguish individual drug types. The anti-resorptive potency of nitrogen-containing bisphosphonates is hundreds (100× for pamidronate, 500× for alendronate) to thousands (2000× for risedronate, 10,000× for zoledronate) of times higher than the first-generation bisphosphonate etidronate [43]. This increased ability to inhibit bone resorption (as opposed to bone mineralization) makes the newer nitrogen-containing bisphosphonates an attractive choice for managing disorders of low bone mass and high bone turnover; these formulations are thus widely used in clinical practice. However, in patients with GACI, the therapeutic goal is to inhibit bone mineralization (as opposed to bone resorption). It is therefore likely that the less commonly used etidronate represents a far better option.
Multiple other factors may influence the choice of bisphosphonate formulation. Etidronate has a low bioavailability at 3% [45], and this can be compounded by malabsorption, which often arises in patients with GACI due to mesenteric ischemia, congestive heart failure, and other factors. At least one case of ulcerative esophagitis due to etidronate has been reported [46], although this appears to be a rare complication [47]. Importantly, lack of availability is perhaps the largest barrier to etidronate’s routine use in patients with GACI, and it is no longer available in the USA as of 2019.
There are no studies available to determine the appropriate duration of treatment. In typical practice, etidronate is often administered for 6–18 months, depending on the degree of vascular calcification on serial imaging studies. Long-term etidronate treatment (in one case for almost 7 years) has been associated with skeletal toxicity [35].
Despite its use in GACI for more than four decades, the clinical benefit of bisphosphonates is far from established and remains an important area of controversy. Bisphosphonates reduce bone remodeling, which may result in increased secondary bone remineralization, raising the theoretical possibility that nitrogen-containing bisphosphonates might exacerbate calcifications [48]. The most informative analysis of the issue included 43 patients who survived GACI beyond the first day of life, with a survival rate of 65% (11/17) in those who received bisphosphonates (given in a variety of formulations) vs. 31% (8/26) in those who did not (p value 0.026, log-rank test) [5]. However, this was a retrospective review, and it excluded the most severely affected patients who had already died by day one of life. Another confounding factor is that patients may exhibit spontaneous resolution of vascular calcifications as part of the natural history of the GACI. It is therefore unknown if the findings in this series reflect the expected natural disease course. There is thus a critical need for prospective studies using consistent, non-nitrogen-containing formulations to define the role of bisphosphonate treatment.
Sodium Thiosulfate
Sodium thiosulfate is an inorganic salt initially developed as an antidote for cyanide poisoning [49]. Based on its supposed chelating effects on calcium salts, it has recently been repurposed for treatment of ectopic calcifications of varying etiologies, including renal failure [50], dermatomyositis [51], hyperphosphatemic tumoral calcinosis [52], and anecdotal reports in patients with GACI [53]. While these small datasets provide preliminarily supportive data, enthusiasm is tempered by potential adverse effects of sodium thiosulfate, including life-threatening metabolic acidosis [54]. Additional studies are therefore needed to determine the safety and efficacy of this approach.
While preliminary data suggests medications such as sodium thiosulfate and bisphosphonates may potentially target vascular calcifications in GACI, importantly, there are no physiologic data to suggest they affect the intimal hyperplasia related to AMP deficiency. This may account for the relatively high mortality that persists in patient with GACI despite these treatments.
Cardiovascular Management
Standard therapy should be initiated for congestive heart failure, pulmonary hypertension, and systemic hypertension. Since renovascular hypertension may result from renal artery stenosis, medications that minimize the rise of angiotensin under renal ischemia, such as angiotensin-converting enzyme (ACE) inhibitors or angiotensin II type 1 receptor blockers (ARBs), are likely to be of benefit. Patients with coronary artery calcification likely benefit from aspirin administration, while patients with profound heart failure may require anticoagulation. If anticoagulation is necessary for severely reduced ejection fraction, it is prudent to avoid the use of warfarin, given its known association with vascular calcification [55]. Heart transplantation was reported in a single individual, who continued to do well with no recurrence of disease over 2 years after the transplant [56]; the authors are also aware of an unpublished patient who continues to do well several years after the heart transplant.
Standard therapy should be entertained for vascular stenosis. As an example, the authors are aware of one patient who required repeated stenting procedures for mesenteric stenosis and ultimately benefitted from a bypass surgical procedure.
Hypophosphatemia Management
The late presentation of hypophosphatemia in patients with GACI raises the interesting possibility that it may ameliorate arterial calcifications and reduce mortality. This concept was first posited by Rutsch et al., who performed a retrospective review of 55 patients and identified an association between hypophosphatemia/hyperphosphaturia and increased survival [5]. There is reasonable physiologic basis to support this speculation: in addition to its phosphaturic effects, FGF23 also exhibits autocrine/paracrine suppression of tissue-nonspecific alkaline phosphatase (TNAP) in a murine model of X-linked hypophosphatemia, leading to local PPi accumulation and impaired skeletal mineralization [57, 58]. Thus, downregulation of TNAP by FGF23 may help preserve circulating PPi and decrease calcification risk in GACI.
However, currently available clinical data is insufficient to support a causal relationship between hypophosphatemia and survival in patients with GACI. An important confounder in the Rutsch et al. series was the high prevalence of early mortality: deaths occurred almost exclusively during the first 6 months, before the development of hypophosphatemia [5]. Hypophosphatemia in both ARHR2 and GACI typically manifest in childhood; it is therefore more likely that survival of the 6-month “critical period” allows hypophosphatemia to become manifest, as opposed to having a direct and/or causative role on mortality. This has important implications for management, as some clinicians avoid treating hypophosphatemia for fear of exacerbating vascular calcifications, placing patients at-risk for potentially disabling musculoskeletal complications. Emerging reports of long-term phosphate replacement without progressive vascular disease suggest that treatment is likely safe [30]. However, longitudinal studies are needed to determine the long-term effects of hypophosphatemia management in patients with GACI.
Hypophosphatemic rickets should be managed with active vitamin D and phosphorus supplementation, as in other causes of FGF23 excess [59]. Typical starting doses include calcitriol at 15 ng/kg/day and phosphorus at 25–30 mg/kg/day divided in 3–4 daily doses. The goal of therapy is not to normalize serum phosphorus (seldom achieved), but rather to correct clinical findings and alkaline phosphatase, while at the same time avoiding the development of medication side effects (such as hypercalciuria from calcitriol or hyperparathyroidism from phosphorus supplementation). Laboratory workup should be performed within 2–3 weeks of initiating treatment or changing medication doses; if this is normal, it can be monitored less frequently. Care should be taken to avoid over-treatment. One patient with GACI developed parenchymal calcification once hypercalciuria ensued from treatment for rickets; however, most of these calcifications (except nephrocalcinosis) regressed after therapeutic doses were adjusted to prevent hypercalciuria [60].
Because hypophosphatemia in ENPP1 deficiency is related to FGF23 excess, one could consider treatment with burosumab, a monoclonal antibody against FGF23. However, there are theoretical reasons to be cautious with this approach. FGF23 is known to downregulate alkaline phosphatase and thus suppress hydrolysis of pyrophosphate [58]. Thus, there is a possibility that burosumab-induced suppression of FGF23 levels could upregulate alkaline phosphatase and increase pyrophosphate hydrolysis, compounding pyrophosphate deficiency and potentially exacerbating calcifications. Of note, the authors are aware of one patient with ENPP1-related rickets who was initially thought to have X-linked hypophosphatemia and received burosumab for months; he did not exhibit any vascular calcification.
Other Management
Monitoring for vascular calcifications is an important component of management in GACI, and there is a critical need for prospective studies to determine appropriate intervals, duration, and modalities. Currently, the authors perform computed tomography imaging every 3–4 months during infancy; this can be performed less frequently (every 6 months in the second year of life) or potentially discontinued if calcifications resolve. Echocardiograms should be performed more frequently depending on the degree of myocardial dysfunction.
Monitoring for extravascular features of GACI should also be considered. This should include routine evaluation for hearing loss starting in infancy. Ophthalmologic monitoring for retinal changes should begin in early childhood. Developmental monitoring and early intervention services should be considered in all infants who survive the critical period.
Future Directions
There is a critical need for targeted therapies to treat vascular disease and prevent early mortality. ENPP1 replacement therapies are an intuitive and exciting potential treatment strategy. In mouse models of ENPP1 deficiency, treatment with ENPP1 bound to the Fc fragment of human IgG1 prevented mortality and vascular calcification [61••] and improved blood pressure and cardiac function [62]. Encouragingly, this targeted approach has the potential to not only prevent vascular calcification, but also intimal hyperplasia [8••] and skeletal complications from hypophosphatemic rickets. A phase 1 trial of ENPP1-Fc is currently in development. The effects of ENPP1-Fc replacement in ABCC6 deficiency are unknown. Additional research is needed to identify the role of ABCC6 in the mineralization cascade in order to develop similarly targeted therapies.
The development of evidence-based treatment strategies has been limited by critical knowledge gaps in understanding the natural history of GACI. In particular, the potential for spontaneous resolution of calcifications in a subset of patients makes it difficult to determine the efficacy of existing and future therapies, because clinicians are at high-risk of assigning causality to interventions that are only correlated with changes in clinical status. Understanding late effects in patients who survive the infant period is also critical to developing effective long-term treatments. Prospective phenotyping studies are needed to define long-term cardiovascular complications, such as arterial stenoses, hypertension, and renovascular disease, which may have important effects on health outcomes and mortality. Phenotyping studies are also needed to better define the spectrum of extravascular involvement.
Conclusions
Conceptions of GACI have evolved dramatically over the past several decades: what was once considered an exclusively infantile arterial disease is now understood as a complex, multisystemic process with a broad phenotypic spectrum. In contrast, advancement in treatment has lagged, in large part due to knowledge gaps in the natural history of GACI that have prevented critical evaluations of treatment outcomes. This has created considerable controversy in the use of commercially available medications (such as bisphosphonates and phosphate replacement therapies) and has prevented the development of standardized treatment guidelines. The possibility of ENPP1 replacement is an exciting prospect offering hope for a targeted, disease-modifying therapy. In that context, prospective, longitudinal natural history studies are one of the highest areas of need and are critical to improving health and well-being for patients with GACI.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Durante G. Athérome congénital de l’aorte et de l’artère pulmonaire. Bull Soc Anat Paris. 1899;74:97–101.
Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19(3):271–3.
Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–54.
Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34(4):379–81.
Rutsch F, Boyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1(2):133–40.
Ruf N, Uhlenberg B, Terkeltaub R, Nurnberg P, Rutsch F. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum Mutat. 2005;25(1):98.
Orriss IR, Arnett TR, Russell RG. Pyrophosphate: a key inhibitor of mineralisation. Curr Opin Pharmacol. 2016;28:57–68.
•• Nitschke Y, Yan Y, Buers I, Kintziger K, Askew K, Rutsch F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. 2018;50(10):–139 This study demonstrated that ENPP1 inhibits neointima formation by generating AMP and that administration of recombinant human ENPP1-Fc protein prevented neointimal formation in an animal model of GACI.
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86(2):267–72.
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86(2):273–8.
Hajjawi MO, MacRae VE, Huesa C, Boyde A, Millan JL, Arnett TR, et al. Mineralisation of collagen rich soft tissues and osteocyte lacunae in Enpp1(−/−) mice. Bone. 2014;69:139–47.
Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90(1):25–39.
Li Q, Brodsky JL, Conlin LK, Pawel B, Glatz AC, Gafni RI, et al. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014;134(3):658–65.
St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364(5):432–42.
Jin H, St Hilaire C, Huang Y, Yang D, Dmitrieva NI, Negro A, et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci Signal. 2016;9(458):ra121.
Chong CR, Hutchins GM. Idiopathic infantile arterial calcification: the spectrum of clinical presentations. Pediatr Dev Pathol : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2008;11(5):405–15.
Witzleben CL. Idiopathic infantile arterial calcification--a misnomer? Am J Cardiol. 1970;26(3):305–9.
Lussier-Lazaroff J, Fletcher BD. Idiopathic infantile arterial calcification: roentgen diagnosis of a rare cause of coronary artery occlusion. Pediatr Radiol. 1973;1(4):224–8.
Attia TH, Abd Alhamed MM, Selim MF, Haggag MS, Fathalla D. Idiopathic arterial calcification of infancy: case report. J Radiol Case Rep. 2015;9(11):32–40.
Shaireen H, Howlett A, Amin H, Yusuf K, Kamaluddeen M, Lodha A. The mystery of persistent pulmonary hypertension: an idiopathic infantile arterial calcification. BMC Pediatr. 2013;13:107.
Sawyer T, Stacey M, Mulreany M, Thompson M, Nitschke Y, Rutsch F, et al. Generalized arterial calcification of infancy associated with meconium peritonitis: a case report and review of the literature. Am J Perinatol. 2009;26(10):711–6.
van der Sluis IM, Boot AM, Vernooij M, Meradji M, Kroon AA. Idiopathic infantile arterial calcification: clinical presentation, therapy and long-term follow-up. Eur J Pediatr. 2006;165(9):590–3.
Glatz AC, Pawel BR, Hsu DT, Weinberg P, Chrisant MR. Idiopathic infantile arterial calcification: two case reports, a review of the literature and a role for cardiac transplantation. Pediatr Transplant. 2006;10(2):225–33.
Galletti S, Nitschke Y, Malavolti AM, Aquilano G, Faldella G, Corvaglia L, et al. Generalized arterial calcification of infancy: fatal clinical course associated with a novel mutation in ENPP1. JIMD Reports. 2011;1:23–7.
Hault K, Sebire NJ, Ho SY, Sheppard MN. The difficulty in diagnosing idiopathic arterial calcification of infancy, its variation in presentation, and the importance of autopsy. Cardiol Young. 2008;18(6):624–7.
Brachet C, Mansbach AL, Clerckx A, Deltenre P, Heinrichs C. Hearing loss is part of the clinical picture of ENPP1 loss of function mutation. Horm Res Paediatr. 2014;81(1):63–6.
Vera J, Lucaya J, Garcia Conesa JA, Aso C, Balaguer A. Idiopathic infantile arterial calcification: unusual features. Pediatr Radiol. 1990;20(8):585–7.
Terry SF, Bercovitch L. Pseudoxanthoma elasticum. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993
Li Q, Schumacher W, Jablonski D, Siegel D, Uitto J. Cutaneous features of pseudoxanthoma elasticum in a patient with generalized arterial calcification of infancy due to a homozygous missense mutation in the ENPP1 gene. Br J Dermatol. 2012;166(5):1107–11.
Ferreira CR, Ziegler SG, Gupta A, Groden C, Hsu KS, Gahl WA. Treatment of hypophosphatemic rickets in generalized arterial calcification of infancy (GACI) without worsening of vascular calcification. Am J Med Genet A. 2016;170a(5):1308–11.
Thumbigere-Math V, Alqadi A, Chalmers NI, Chavez MB, Chu EY, Collins MT, et al. Hypercementosis associated with ENPP1 mutations and GACI. J Dent Res. 2018;97(4):432–41.
Staretz-Chacham O, Shukrun R, Barel O, Pode-Shakked B, Pleniceanu O, Anikster Y, et al. Novel homozygous ENPP1 mutation causes generalized arterial calcifications of infancy, thrombocytopenia, and cardiovascular and central nervous system syndrome. Am J Med Genet A. 2019;179(10):2112–8.
Dlamini N, Splitt M, Durkan A, Siddiqui A, Padayachee S, Hobbins S, et al. Generalized arterial calcification of infancy: phenotypic spectrum among three siblings including one case without obvious arterial calcifications. Am J Med Genet A. 2009;149a(3):456–60.
Cheng KS, Chen MR, Ruf N, Lin SP, Rutsch F. Generalized arterial calcification of infancy: different clinical courses in two affected siblings. Am J Med Genet A. 2005;136(2):210–3.
Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, Kiffer-Moreira T, et al. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res. 2013;28(2):419–30.
Ciana G, Trappan A, Bembi B, Benettoni A, Maso G, Zennaro F, et al. Generalized arterial calcification of infancy: two siblings with prolonged survival. Eur J Pediatr. 2006;165(4):258–63.
Thomas P, Chandra M, Kahn E, McVicar M, Naidich J, LaCorte M. Idiopathic arterial calcification of infancy: a case with prolonged survival. Pediatr Nephrol (Berlin, Germany). 1990;4(3):233–5.
Ciana G, Colonna F, Forleo V, Brizzi F, Benettoni A, de Vonderweid U. Idiopathic arterial calcification of infancy: effectiveness of prostaglandin infusion for treatment of secondary hypertension refractory to conventional therapy: case report. Pediatr Cardiol. 1997;18(1):67–71.
Sholler GF, Yu JS, Bale PM, Hawker RE, Celermajer JM, Kozlowski K. Generalized arterial calcification of infancy: three case reports, including spontaneous regression with long-term survival. J Pediatr. 1984;105(2):257–60.
Marrott PK, Newcombe KD, Becroft DM, Friedlander DH. Idiopathic infantile arterial calcification with survival to adult life. Pediatr Cardiol. 1984;5(2):119–22.
Thiaville A, Smets A, Clercx A, Perlmutter N. Idiopathic infantile arterial calcification: a surviving patient with renal artery stenosis. Pediatr Radiol. 1994;24(7):506–8.
Meradji M, de Villeneuve VH, Huber J, de Bruijn WC, Pearse RG. Idiopathic infantile arterial calcification in siblings: radiologic diagnosis and successful treatment. J Pediatr. 1978;92(3):401–5.
Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032–45.
Stuart AG. Idiopathic arterial calcification of infancy and pyrophosphate deficiency. J Pediatr. 1993;123(1):170–1.
Etidronate [package insert]. Cincinatti, OH: Procter & Gamble Pharmaceuticals. 2009.
Macedo G, Azevedo F, Ribeiro T. Ulcerative esophagitis caused by etidronate. Gastrointest Endosc. 2001;53(2):250–1.
van Staa T, Abenhaim L, Cooper C. Upper gastrointestinal adverse events and cyclical etidronate. Am J Med. 1997;103(6):462–7.
Boivin G, Farlay D, Bala Y, Doublier A, Meunier PJ, Delmas PD. Influence of remodeling on the mineralization of bone tissue. Osteoporos Int. 2009;20(6):1023–6.
Reade MC, Davies SR, Morley PT, Dennett J, Jacobs IC. Review article: management of cyanide poisoning. Emerg Med Australas. 2012;24(3):225–38.
Adirekkiat S, Sumethkul V, Ingsathit A, Domrongkitchaiporn S, Phakdeekitcharoen B, Kantachuvesiri S, et al. Sodium thiosulfate delays the progression of coronary artery calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25(6):1923–9.
Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr. 2012;160(3):520–2.
Jost J, Bahans C, Courbebaisse M, Tran TA, Linglart A, Benistan K, et al. Topical sodium thiosulfate: a treatment for calcifications in Hyperphosphatemic familial tumoral calcinosis? J Clin Endocrinol Metab. 2016;101(7):2810–5.
Omarjee L, Nitschke Y, Verschuere S, Bourrat E, Vignon MD, Navasiolava N, et al. Severe early-onset manifestations of pseudoxanthoma elasticum resulting from the cumulative effects of several deleterious mutations in the ENPP1, ABCC6 and HBB genes: transient improvement in ectopic calcification with sodium thiosulfate. Br J Dermatol 2019.
Selk N, Rodby RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24(1):85–8.
Poterucha TJ, Goldhaber SZ. Warfarin and vascular calcification. Am J Med. 2016;129(6):635.e1–4.
Giovannoni I, Callea F, Travaglini L, Amodeo A, Cogo P, Secinaro A, et al. Heart transplant and 2-year follow up in a child with generalized arterial calcification of infancy. Eur J Pediatr. 2014;173(12):1735–40.
Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive Osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp mice. PLoS Biol. 2016;14(4):e1002427.
Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, et al. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis. 2019;14(1):58.
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13–30.
Freychet C, Gay C, Lavocat MP, Teyssier G, Patural H, Bacchetta J, et al. GACI syndrome: a case report with a neonatal beginning. Arch Pediatr. 2014;21(6):632–6.
•• Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. 2015;6:10006 This study demonstrated that administration of a recombinant ENPP1-Fc fusion protein prevented vascular calcifications and mortality in an animal model of GACI.
Khan T, Sinkevicius KW, Vong S, Avakian A, Leavitt MC, Malanson H, et al. ENPP1 enzyme replacement therapy improves blood pressure and cardiovascular function in a mouse model of generalized arterial calcification of infancy. Dis Model Mech. 2018;11(10):dmm035691.
Funding
This research was supported by the Intramural Research Programs of the NIDCR and NHGRI, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The NIH receives funding from Inozyme for research related to GACI. The NIDCR receives research funding from Amgen and QED, outside the submitted work. Dr. Gafni serves as an advisory board member to Ascendis and an unpaid member of the medical advisory board for GACI Global.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Rare Bone Disease
Rights and permissions
About this article

Cite this article
Boyce, A.M., Gafni, R.I. & Ferreira, C.R. Generalized Arterial Calcification of Infancy: New Insights, Controversies, and Approach to Management. Curr Osteoporos Rep 18, 232–241 (2020). https://doi.org/10.1007/s11914-020-00577-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-020-00577-4