
Abstract
The bone is a common site for metastasis in patients with advanced prostate carcinoma, and provides a ‘fertile’ milieu which stimulates tumour growth and associated bone disease. For years, the concept of treatment strategies has remained targeting the tumour itself; however, the occurrence of chemoresistance remains a challenge now more than ever. The attraction of targeting the bone microenvironment in order to disrupt tumour localisation and proliferation stems from the idea that stromal cells are superiorly stable at a genetic level, thus decreasing the risk of resistance manifestation. In this review, we will discuss recent findings with regards to the pathogenesis of prostate cancer-induced bone disease and recent therapeutic strategies in an aim to evaluate the ever increasing role of the microenvironment in disease progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is the most common form of cancer amongst men in the UK. Skeletal metastasis is a frequent complication of castration-resistant disease causing considerable morbidity. On average, a patient with metastatic disease will experience a skeletal-related event every 3 to 6 months. However, occurrences of this nature are not regular, with frequency of events increasing with cancer progression. As the disease becomes more extensive, treatment options are reduced and reliability of therapeutics decreased [1]. Cancer invasion and metastasis mark the transformation of a locally growing tumour into a systemic, metastatic, life-threatening disease [2].
Metastatic cancer cells produce factors that modulate normal bone remodelling, giving rise to both osteoblastic and osteolytic lesions. Symptoms of patients that have developed cancer-induced bone disease include the following: extreme bone pain, hypercalcemia, pathological fractures, and in some cases, spinal cord and nerve compression [3]. Bone metastasis is a common complication amongst many progressive solid tumour types. Less than 20 % of patients, however, will survive for more than 5 years after the discovery of cancer-induced bone disease [4].
Understanding the patterns of metastasis has historically been and still remains a challenge. In the early twentieth century, two theories were formulated in a bid to explain the specific metastatic patterns of certain tumour types: the ‘mechanical’ and the ‘seed-and-soil’ hypotheses. The mechanical hypothesis predicts metastasis outcome by the spread of the primary tumour into the lymphatic system, subsequently resulting in its spread through the venous system, whereas the seed-and-soil hypothesis describes metastasis as a plant going to seed. The seeds can be carried in all directions, but will only survive if they fall on ‘congenial soil’ [5]. Since then, there have been many valuable contributions to the understanding of cancer pathogenesis, metastasis and the dependency of this process on the crosstalk between the tumour and the cancer-microenvironment. Gundem et al. recently showed very clearly, using whole genome sequencing, the evolution of metastatic prostate cancer from initial tumorigenesis through to metastasis and castration resistance [6••]. This work indicates that subclones within the primary tumour develop metastatic potential from the beginning of the disease, rather than the primary tumour developing this metastatic potential as a whole. The pattern of metastatic spread was also investigated, identifying that tumour cells frequently spread from one metastatic site to another. This study lends support to the ‘seed-and-soil’ hypothesis in that rare subclones develop metastatic potential within the primary tumour and are able to give rise to disease progression as a result of environmental changes. The now widely known concept of the ‘vicious cycle’ first proposed by Mundy et al. eloquently explains how cancer cells are able to manipulate their immediate environment to support their survival and growth [7]. This has been followed by numerous studies investigating the interactions between the tumour and bone marrow microenvironment and thus therapeutic strategies aimed to exploit those findings.
Homing to the Bone and the Pre-metastatic Niche
The initial stages of metastasis involve the detachment of malignant cells from the primary tumour and migration of these cells into nearby vasculature. In the normal prostate gland, cells have restricted migratory capacity. Cell-to-cell adhesion is maintained by a complex of cell adhesion molecules such as selectins and cadherins. Early in the process of migration, prostate cancer cells exhibit alterations in the expression of different molecules that lead to decreased cellular adhesion. The process of epithelial-to-mesenchymal transition (EMT) is now regarded by many to be critical in the development of more migratory and invasive tumour types [8]. However, controversially in 2015, two independent studies challenged the traditional role of EMT in metastasis. Zheng et al. showed that in vivo knockout of either twist1 or snail1, two key transcription factors responsible for EMT, did not alter either progression of pancreatic cancer or the capacity of local invasion or metastasis [9•]. Fischer et al. found that lung metastases were comprised primarily of tumour cells that maintained their epithelial phenotype and had not undergone EMT. Notably, both studies identified a potential role for EMT in chemoresistance [10•]. Since prostate cancer bone metastasis is traditionally thought to be dependent upon EMT for the early stages of the metastatic cascade, it is intriguing to speculate that this role for EMT may not be as significant as first thought.
Once cells intravasate, the initial attraction of detached cells to distal sites is largely regulated by a series of integrins and chemokines produced by the bone marrow and stromal cells [11]. Amongst these, stromal-derived factor-1 (SDF-1), also known as C-X-C chemokine ligand 12 (CXCL12) is thought to play a major role. The receptor for CXCL12, C-X-C chemokine receptor 4 (CXCR4), is present on osteoclast precursors and regulates haematopoietic cells homing to bone [12]. Like haematopoietic stem cell (HSC) precursors, cancer cells also express CXCR4 and are thus attracted into the bone microenvironment [13]. At a time when the CXCL12/CXCR4 axis was beginning to be recognised as a modulator of migration and survival in many malignant cell types, Sun et al. showed data for prostate cancer, supporting the concept that CXCL12 and CXCR4 expression is associated with a progressive cancer type [14]. In addition to producing large amounts of CXCL12, osteoblasts also express anchorage molecules such as angiopoietin (Ang-1) and osteopontin (OPN) that also encourage tumour cells into the bone microenvironment. OPN is a glycophosphoprotein with the ability to stimulate HSC and osteoclast adherence to bone matrix. It has a key role in the trans-marrow migration, retention and negative regulation of HCSs within the osteoblastic niche. Furthermore, it has been shown in both breast and prostate cancer that OPN is linked to regulation of metastatic spread and has been found to be highly expressed both within metastatic cells and surrounding stromal tissue [15].
Dormancy and the HSC Niche
HSCs also have the ability to engage in a reversible state of cell cycle arrest, termed ‘quiescence’. Quiescence allows HSCs to escape damage by cellular toxins and stresses, thus maintaining a viable stem cell reserve. In a similar way, disseminated tumour cells (DTCs) also share mechanisms of ‘dormancy’. Dormancy is thought to allow tumour cells to evade cell death from chemotherapeutics. Traditional chemotherapy works by targeting rapidly dividing cells. By engaging reversible cell cycle arrest, however, cancer cells become resistant to these effects. Bone metastatic DTCs target the osteoblastic haematopoietic stem cell niche via the CXCL12/CXCR4 axis and compete for occupancy of the niche. Upon binding to the niche, tumour cells are thought to undergo growth arrest resulting in dormancy [16, 17]. Recent evidence also suggests that quiescent cells are more tumourigenic in a murine model of bone metastasis, when compared with rapidly dividing cells, providing further support for the importance of such tumour cells [18]. One molecule thought to be implicated in homing to the HSC niche is annexin II. Annexin II is expressed on the surface of osteoblasts and, in conjunction with its receptor, has the ability to regulate homing to bone in a similar fashion to the CXCL12/CXCR4 interaction [19]. Shiozawa et al. showed that by blocking annexin II or its receptor in animal models of prostate cancer, short- and long-term localisation of cancer cells could be limited [20]. Thus current evidence suggests that prostate cancer is able to utilise HSC homing mechanisms in order to invade and localise within the bone marrow niche. This raises the possibility that approaches which mobilise stem cells from the HSC niche, such as the CXCR4 inhibitor AMD3100, may also mobilise tumour cells and so render them susceptible to chemotherapy [16, 17].
The Tumour-Bone Microenvironment
The term ‘bone microenvironment’ broadly describes the complex biological interplay between cells of the haematopoietic and mesenchymal origin, the bone marrow stroma and the bone extracellular matrix. The bone matrix serves as a major source of growth factors, including tumour growth factor-β (TGF-β), insulin growth-like factors (IGFs), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and bone morphogenetic proteins (BMPs). Collectively, these embedded growth factors make the bone matrix an attractive site for metastasis, enabling the growth of the metastatic tumour in situ and increasing the production and release of cytokines and other bone remodelling factors from the tumour itself [21].
Osteoblasts
Normal bone development is regulated by complex interactions between cells of the bone microenvironment including osteoblasts, which control bone formation and osteoclasts, which control bone resorption. Cancer cells use the same regulatory pathways that are involved in normal bone development and remodelling in order to ‘hijack’ bone turnover. The dysregulation of bone remodelling seen in cancer-induced bone metastasis is the result of the interactions between the tumour cells and the stromal cells of the bone marrow microenvironment.
Osteoblast activation and maturation in multiple myeloma, breast and prostate cancer has been shown to be stimulated by the wnt pathway [22]. Accumulating evidence suggests that wnt released by metastatic prostate cancer cells can stimulate osteoblasts and enhance tumour proliferation, whilst the inhibitor of Wnt signalling, dickkopf-1 (DKK1) can promote osteolysis, particularly during the early stages of cancer development. Expression of DKK1 has been shown to be upregulated in early developing prostate cancer with a decline in DKK1 levels occurring in advanced bone metastases. This suggests that the initial upregulation of DKK1 is required for the establishment of the tumour, whereas the DKK1 decrease during bone metastasis can promote wnt expression and thereby result in an increase in osteoblast activity to give rise to the osteoblastic metastases traditionally associated with prostate cancer [23, 24].
Other paracrine factors secreted by prostate cancer that regulate osteoblast proliferation and/or differentiation include the following: BMP, TGF-β, IGF, PDGF, vascular endothelial growth factor (VEGF) and endothelin-1 (ET-1) [25]. Numerous members of the TGF-β family have been found to stimulate bone formation. Serum TGF-β concentrations in prostate cancer patients have been found to be elevated in bone metastatic compared with non-bone metastatic patients [26]. Another study showing the importance of one of these factors is the work by Autzen et al. who found that expression of BMP6 mRNA was upregulated in primary bone metastatic prostate cancer samples, suggesting that BMPs and specifically BMP-6 could potentially play a role in the mediation of skeletal metastasis [27].
VEGF has previously been shown to regulate bone formation by controlling vascularity within the developing growth plate and has been shown by Street et al. to differentiate primary osteoblasts in vitro, thus suggesting that VEGF may enhance bone formation and repair [28].
Osteoclasts
Osteoclasts function primarily as mediators of bone resorption and maintain bone homeostatic balance through continual remodelling of the microenvironment in response to various stimuli [29]. Under normal physiological conditions, osteoblastic cells regulate osteoclast activity by maintaining a fine balance between osteoprotegerin (OPG) and receptor activator of nuclear factor k-B ligand (RANKL) expression. The binding of RANKL to receptor activator of nuclear factor k-B (RANK) on osteoclast precursors initiates a signalling cascade resulting in activation and differentiation of osteoclasts. The interaction was shown to be an absolute requirement for signalling as early as 1999 by Dougall et al., [30] who demonstrated that both whole body RANKL−/− and RANK−/− mice developed abnormally dense bones due to the absence of osteoclasts. Dysregulation of RANKL through the secretion of parathyroid hormone-related protein (PTHrP) has become a typically described mechanism in osteolytic metastasis in breast cancer. RANKL binds to RANK on osteoclast precursors and stimulates the expression of genes such as integrin αvβ3, cathepsin K, matrix metallopeptidase 9 (MMP-9) and H+-ATPase necessary for osteoclast adhesion to the bone and bone degradation. The degradation of bone then promotes the proliferation of tumour cells through the release of growth factors. These growth factors then stimulate the proliferation of both osteoblasts and tumour cells to create a vicious cycle [22]. The dysregulation of the OPG/RANKL and/or RANK crosstalk has been shown to occur in a number of cancers, with the production of soluble RANKL in prostate cancer suggested as a mechanism by which osteoclastogenesis may be initiated [31].
Bone Marrow-Derived Adipocytes
Bone is a major regulator of energy metabolism. Adipocytes and osteoblasts share common precursors known as mesenchymal stem cells (MSCs). Adipogenic differentiation plays an important role in regulating bone mass and homeostasis, as cells of the MSC lineage can be diverted towards the adipogenic or osteoblastic lineage depending on the presence of adipogenic (e.g. peroxisome proliferator-activated receptor-γ (PPARγ)) or osteogeneic (e.g. runt-related transcription factor 2 (Runx2), core-binding factor alpha 1 (Cbfa1)) factors that may be present in the bone microenvironment. A number of studies have suggested that the behaviour of tumour cells can be affected by the presence of adipocytes and adipocyte-associated factors. An increase in adipocyte number can lead to an abundance of lipids, which are critical for signalling, cellular trafficking and migration. Transformed cells have the ability to utilise and store lipids in order to gain a growth advantage compared to normal epithelial cells. Moreover, adipocytes and associated inflammatory cells secrete adipokines and cytokines, which are known to contribute towards tumour proliferation and survival [32]. A recent study has shown a functional relationship between bone marrow adipocytes and metastatic prostate cancer. Herroon et al., found that an increase in invasion was observed in prostate cancer cells exposed to adipocyte-conditioned media. This increased invasion was found to be mediated by fatty acid-binding protein 4 (FABP4), suggesting that the presence of adipocyte-related factors can give rise to a more progressive phenotype [33]. Furthermore, marrow adipocytes have been found to secrete C-X-C chemokine ligand 1 (CXCL1) and C-X-C chemokine ligand 2 (CXCL2), chemokines postulated to activate osteoclasts, thereby providing a potential mechanism linking marrow adipocytes to the vicious cycle of cancer-induced bone disease [34].
Bone Marrow Mesenchymal Stem Cells and Stromal Cells
Several studies have shown that bone marrow mesenchymal stem cells (MSCs) have the potential to promote the progression of various tumour types. Due to the multipotent nature of this lineage, MSCs can give rise to a variety of cell types including the following: osteoblasts, adipocytes, chondrocytes and fibroblasts [35]. MSCs are known to be recruited to the primary tumour site to facilitate tumour progression and metastasis. Jung et al. provide evidence that the recruitment of MSCs to prostate cancer is dependent on the expression of C-X-C chemokine receptor 6 (CXCR6). CXCR6 signalling was shown to support recruitment, conversion and activation of MSCs into CXCL12-secreting cancer-associated fibroblasts (CAFs) [36••]. CAFs are well known to play essential roles in primary tumour growth and metastasis, and their crucial role in tumour growth within the bone environment is beginning to emerge [37]. Li et al. showed that expression of TGF-β type II receptor was lost in prostate CAFs, suggesting that the loss of stromal TGF-β responsiveness in the primary site promoted prostate cancer mixed bone metastasis, which was found to be mediated through an increase in the expression of cytokines such as CXCL1 and C-X-C chemokine ligand 16 (CXCL16) [38].
Therapeutic Strategies for Targeting the Tumour-Bone Microenvironment
Androgen deprivation therapy combined with surgery remains the first line of treatment for localised prostate cancer. However, with 90 % of patients with castration-resistant disease developing bone metastasis, the effects of primary treatment become more sinister. Androgen deprivation therapy is known to cause bone loss and in 2014, Ottewell et al. demonstrated, with the use of in vivo models, that mimicking androgen ablation increased growth of bone disseminated tumour through osteoclast-mediated mechanisms [39]. As targeting resistant tumour cells remains a considerable challenge, efforts to target the tumour microenvironment present a unique advantage. Stromal cells are far more genetically stable compared to tumour cells and are therefore less susceptible to the possibility of therapeutic resistance. Furthermore, the diversity of tumour-stroma crosstalk that contributes towards cancer progression within the bone microenvironment provides a wide range of potential therapeutic targets [40].
Bisphosphonates
Bisphosphonates are chemically stable derivatives of inorganic pyrophosphate that inhibit calcification by binding to bone mineral, preventing its breakdown by osteoclasts. Second-generation bisphosphonates now have an added effect of inhibiting mevalonate pathway enzymes, which also have direct effects on osteoblasts and tumour cells. These compounds can block apoptosis and promote differentiation of osteoblasts and also promote apoptosis and inhibit invasion of tumour cells [41]. Long-term clinical trials in patients with metastatic hormone-refractory prostate cancer have shown that treatment with the most commonly used bisphosphonate in prostate cancer, zoledronic acid, reduced overall risk of skeletal complications by 36 % [42, 43].
Bisphosphonates are generally well tolerated and have beneficial effects on the management of metastatic bone disease and the prevention of treatment-induced bone loss. However, despite obvious clinical benefits, it is clear that only a proportion of skeletal complications are prevented with bisphosphonates and increases in overall survival in patients with cancers metastatic to the skeleton has yet to be achieved [44].
Targeting the RANKL/RANK/OPG interaction
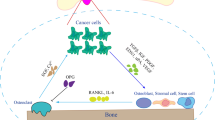
Signalling of the RANKL/RANK/OPG triad has been shown to have significant involvement in bone metastasis from both breast and prostate cancer. Blocking the RANKL/RANK interaction has shown a unique anti-tumour effect within the bone [45]. Denosumab is a humanised monoclonal antibody that binds to and neutralises RANKL, thereby inhibiting osteoclast function and subsequent bone resorption (Fig. 1). Phase 3 studies in men with castration-resistant prostate cancer showed denosumab to significantly increase bone metastasis-free survival and delay onset of bone metastasis. However, no increase in overall survival was seen [46]. Phase 3 studies comparing denosumab to zoledronic acid in patients suffering from breast, prostate cancer or multiple myeloma suggested that the effect of denosumab on skeletal-related events was superior to those of zoledronic acid. Denosumab delayed the onset of skeletal-related events by 8.21 months compared to zoledronic acid. Overall survival, however, was found to be similar between treatment groups [47].

Schematic representation of therapeutic targets in the tumour-bone microenvironment. (1) Targeting VEGF. Bevacizumab binds directly to VEGF, inhibiting activity. Sunitinib and/or sorafenib, multiple receptor tyrosine kinase inhibitors, prevent VEGFR binding. IMS-1121B, an anti-VEGFR2 human monoclonal antibody, prevents VEGF binding. (2) Factors stimulating osteoblastic activity. TGF-β, FGF, PDGF, IGF, ET-1, BMPs and wnt all stimulate osteoblast activity. ET-1 signalling inhibits DKK1 and thus increases wnt. Atrasentan is an ET-1 inhibitor, preventing the inhibition of DKK1, a negative regulator of wnt signalling. (3) Targeting RANKL. Denosumab, a humanised monoclonal antibody, binds to RANKL, thereby inhibiting osteoclast activity and thus osteoclastogenesis. (4) Targeting osteoclasts. Bisphosphonates, such as zoledronic acid, inhibit mevalonate pathway enzymes and bind to bone mineral to prevent its resorption via osteoclasts. (5) Bone-targeted radiopharmaceuticals. Radium-223 causes double stranded DNA breaks in cancer cells. Short-alpha particle paths allow effective localised cytotoxic cell death with reduced off-target effects
Targeting VEGF Signalling
VEGF has been found in many tumour types to exert a driving role in tumour angiogenesis, growth, invasion and metastasis. VEGF is expressed by osteoblasts and promotes chemotactic migration, proliferation and differentiation effects on osteoblasts, as well as stimulating the formation and survival of osteoclasts. The actions of VEGF are thought to contribute to tumour cell recognition of the bone and establishment of tumour cells within the skeleton. Increased VEGF expression has been associated with a more aggressive phenotype in castration-resistant prostate cancer. With therapies targeting the VEGF pathway showing promising early clinical application, these inhibitors are now being investigated in clinical trials [48].
Bevacizumab was the first agent to provide clinical evidence that the use of VEGF inhibitors to target the microenvironment may provide patient benefits. Bevacizumab was FDA approved in 2004 primarily for the treatment of metastatic colon cancer, but has since been used to target other metastatic cancers in combination with cytotoxic agents.
In phase 2 studies, the combination of bevacizumab and docetaxel in hormone-refractory prostate cancer patients showed promising results. These results, however, did not transpire to a phase 3 study where despite a small improvement in progression free survival, overall survival did not improve [49, 50].
Emerging evidence now suggesting suggests that VEGF inhibitors may increase delivery of chemotherapeutics by increasing blood flow to the tumour itself. In the majority of cancers, tumour-associated blood vessels are often abnormal in both structure and function. Abnormal tumour vessels can impede the function of immune cells as well as the transport of chemotherapy and oxygen. Vascular normalisation is a therapeutic strategy aimed at enhancing treatment delivery through the remodelling of tumour vessels in order to partially overcome physiological barriers that prevent effective chemotherapeutic activity. This approach, unfortunately, appears to be both time and dose dependent, with a narrow window of opportunity to increase blow flow that may well differ between cancer types [51]. Accordingly, the studies of Di Lorenzo and Kelly et al., in which combining bevacizumab was combined with docetaxel, demonstrate this reality, showing a small improvement but without achieving a significant effect on overall survival. Given the difficulty of implementing this strategy, Wong et al. have recently proposed an alternative approach termed ‘vascular promotion therapy.’ Using co-administration of low-dose cilengitide, an angiogenesis inhibitor and verapamil, a calcium channel blocker, Wong et al. were able to show increased vessel dilation and blood flow in both mouse and human cancer models. This approach was associated with increased treatment delivery of gemcitabine, with a resultant reduction in tumour growth and metastasis, along with minimal side effects and an increase in overall survival. These data demonstrates the potential of VEGF inhibitors in combination with other agents to improve efficacy of chemotherapeutics [52].
Other VEGF inhibitors currently being evaluated include the following: small molecule receptor tyrosine kinase inhibitors, sunitinib and sorafenib and receptor-specific antibodies IMC-1121B and anti-VEGFR-2 (Fig. 1). Sunitinib and sorafenib have already been approved by the FDA for the treatment of advanced renal cell carcinoma and are currently being investigated as therapies for other progressive cancer types including castration-resistant prostate cancer [53]. These data demonstrate the potential of VEGF inhibitors for targeting the tumour microenvironment and suggest the possibility of future combination therapies to improve chemotherapeutic efficacy.
Endothelin-1 Inhibitors
ET-1 has been implicated as having a central role in the mediation of osteosclerotic metastasis, as ET-1 stimulates bone formation and osteoblast proliferation [54]. One way ET-1 regulates osteoblast function is by inhibition of DKK1 in marrow stromal cells, thus increasing Wnt signalling (Fig. 1). Furthermore, preclinical data has provided evidence that by blocking the ET-1 receptor, osteosclerotic lesion occurrence can be prevented. Serum levels of ET-1 have been shown to be elevated in prostate cancer patients with bone metastases. Despite promising initial findings, however, subsequent phase 3 trials evaluating the potential of endothelin receptor antagonists, such as atrasentan, have failed to provide clinically significant benefits to patients with prostate cancer [55].
Bone-Targeted Radiopharmaceuticals
Radium-223 dichloride (radium-223) is an alpha emitter that selectively binds to areas of increased bone turnover, a characteristic traditionally associated with the osteoblastic metastases of prostate cancer. By inducing double-strand DNA breaks, radium-223 treatment results in a potent and highly localised cytotoxic effect in the target areas (Fig. 1). The short path of the alpha particles also means that off-target toxic effects in the surrounding tissue can be reduced. In both phase 1 and 2 studies, radium-223 has shown a favourable safety profile, with minimal myelotoxicity. Furthermore, phase 2 studies have also observed a reduction in bone pain and an improvement in disease-related biomarkers (e.g. serum levels of alkaline phosphatase and prostate-specific antigen (PSA)). In the pivotal phase 3 radium-223 in symptomatic prostate cancer patients (ALSYMPCA) study conducted by Parker et al. in 2013, radium-223 significantly prolonged overall survival in patients with castration-resistant prostate cancer and bone metastases, with a overall 30 % reduction in the risk of death [56]. A number of trials are now beginning to look at combining radium 223 with other agents. A phase 1/2 study of the efficacy of radium-223 with docetaxel (NCT01106352) has recently been completed, and results are pending. In addition, a phase 2 trial investigating the combination of radium-223 with abiraterone or enzalutamide (NCT02034552), and a phase 3 trial combining radium-223 with abiraterone and prednisone (NCT02043678) are ongoing [57].
Conclusion
Therapeutic strategies for bone metastasis are driven by our evolving understanding of the molecular interactions between cancer cells and the bone microenvironment. Although recent clinical studies of targetable agents have in many ways been less then successful, many illustrate the potential for bone microenvironment manipulations as a treatment option for cancer-induced bone disease. The potential for targeting the tumour microenvironment in combination with chemotherapeutics remains a largely unexplored area. With recent findings demonstrating that the inhibition of many processes associated with advanced disease significantly improves the efficacy of treatment, this is bound to become a rapidly expanding area of research. As such, further elucidation of the complex interplay between cancer cells and stroma within the metastatic niche will undoubtedly bring to light new avenues for therapeutic strategies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Coleman RE, 20 Pt 2. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–9.
Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009.
Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350(16):1655–64.
Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–25.
Hess KR et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106(7):1624–33.
Gundem G et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–7. Uses whole genome sequencing to describe the pattern of metastatic spread in advanced prostate cancer.
Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–93.
Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer. 2011;128(11):2545–61.
Zheng X et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–30. Suggests that EMT is not critical for metastasis.
Fischer KR et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6. Suggests that EMT is not critical for metastasis.
Autio KA, Morris MJ. Targeting bone physiology for the treatment of metastatic prostate cancer. Clin Adv Hematol Oncol: H&O. 2013;11(3):134–43.
Buenrostro D, Park SI, Sterling JA. Dissecting the role of bone marrow stromal cells on bone metastases. Biomed Res Int. 2014;2014, 875305.
Taichman RS et al. The evolving biology and treatment of prostate cancer. J Clin Invest. 2007;117(9):2351–61.
Sun YX et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89(3):462–73.
Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev. 2006;25(4):521–9.
Shiozawa Y et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–312.
Wang N et al. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models. J Bone Miner Res. 2014;29(12):2688–96.
Wang N et al. Mitotic quiescence, but not unique “stemness,” marks the phenotype of bone metastasis-initiating cells in prostate cancer. FASEB J. 2015;29(8):3141–50.
Pedersen EA et al. The prostate cancer bone marrow niche: more than just ‘fertile soil’. Asian J Androl. 2012;14(3):423–7.
Shiozawa Y et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem. 2008;105(2):370–80.
Zheng Y et al. The role of the bone microenvironment in skeletal metastasis. J Bone Oncol. 2013;2(1):47–57.
Chantrain CF et al. Bone marrow microenvironment and tumor progression. Cancer Microenviron. 2008;1(1):23–35.
Hall CL et al. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65(17):7554–60.
Hall CL et al. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97(4):661–72.
Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5(1):21–8.
Shariat SF et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol. 2001;19(11):2856–64.
Autzen P et al. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br J Cancer. 1998;78(9):1219–23.
Street J et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci U S A. 2002;99(15):9656–61.
Sottnik JL, Keller ET. Understanding and targeting osteoclastic activity in prostate cancer bone metastases. Curr Mol Med. 2013;13(4):626–39.
Dougall WC et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412–24.
Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. 2004;91(4):718–29.
Hardaway AL et al. Bone marrow fat: linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014;33(2–3):527–43.
Herroon MK et al. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget. 2013;4(11):2108–23.
Hardaway AL et al. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis. 2015;32(4):353–68.
Luo J et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene. 2014;33(21):2768–78.
Jung Y et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. Demonstrates a role for CXCR16 in recruiting MSCs into prostate tumours and promoting metastasis.
Olechnowicz SW, Edwards CM. Contributions of the host microenvironment to cancer-induced bone disease. Cancer Res. 2014;74(6):1625–31.
Li X et al. Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol Cancer Res. 2012;10(4):494–503.
Ottewell PD et al. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr Relat Cancer. 2014;21(5):769–81.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37.
Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8(6):357–68.
Saad F et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94(19):1458–68.
Saad F et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879–82.
Coleman RE, McCloskey EV. Bisphosphonates in oncology. Bone. 2011;49(1):71–6.
Ando K et al. RANKL/RANK/OPG: key therapeutic target in bone oncology. Curr Drug Discov Technol. 2008;5(3):263–8.
Smith MR et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379(9810):39–46.
Lipton A et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48(16):3082–92.
Roberts E, Cossigny DA, Quan GM. The role of vascular endothelial growth factor in metastatic prostate cancer to the skeleton. Prostate Cancer. 2013;2013:418340.
Di Lorenzo G et al. Combination of bevacizumab and docetaxel in docetaxel-pretreated hormone-refractory prostate cancer: a phase 2 study. Eur Urol. 2008;54(5):1089–94.
Kelly WK et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J Clin Oncol. 2012;30(13):1534–40.
Huang D et al. Anti-angiogenesis or pro-angiogenesis for cancer treatment: focus on drug distribution. Int J Clin Exp Med. 2015;8(6):8369–76.
Wong PP et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell. 2015;27(1):123–37.
Fang H, DeClerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73(16):4965–77.
Muralidharan A, Smith MT. Pathobiology and management of prostate cancer-induced bone pain: recent insights and future treatments. Inflammopharmacology. 2013;21(5):339–63.
Todenhofer T et al. Targeting bone metabolism in patients with advanced prostate cancer: current options and controversies. Int J Endocrinol. 2015;2015:838202.
Parker C et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213–23.
Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85(4):717–24.
Acknowledgments
This work was supported by the Prostate Cancer UK and Cancer Research UK (CR-UK) grant number C38302/A12278, through the Oxford Cancer Research Centre Development Fund and through the University of Oxford Medical Research Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Christina J. Turner and Claire M. Edwards declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Osteoporosis and Cancer
Rights and permissions
About this article

Cite this article
Turner, C.J., Edwards, C.M. The Role of the Microenvironment in Prostate Cancer-Associated Bone Disease. Curr Osteoporos Rep 14, 170–177 (2016). https://doi.org/10.1007/s11914-016-0323-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-016-0323-2