
Abstract
Synovial joint morphogenesis occurs through the condensation of mesenchymal cells into a non-cartilaginous region known as the interzone and the specification of progenitor cells that commit to the articular fate. Although several signaling molecules are expressed by the interzone, the mechanism is poorly understood. For treatments of cartilage injuries, it is critical to discover the presence of joint progenitor cells in adult tissues and their expression gene pattern. Potential stem cell niches have been found in different joint regions, such as the surface zone of articular cartilage, synovium, and groove of Ranvier. Inherited joint malformations as well as joint-degenerating conditions are often associated with other skeletal defects and may be seen as the failure of morphogenic factors to establish the correct microenvironment in cartilage and bone. Therefore, exploring how joints form can help us understand how cartilage and bone are damaged and develop drugs to reactivate this developing mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Formation and positioning of synovial joints are critically important during evolution, allowing successful adaptation of vertebrate limbs to a variety of ecological environments. Because of the variety of tissues comprising the synovial joint (articular cartilage, synovial fluid, ligaments, joint capsule), disease or trauma affecting one tissue has wide-ranging implications on the health and function of the joint as a whole organ [1]. Inherited joint malformations, although relatively rare, present with a wide range of forms and severity and are often associated with skeletal defects, suggesting a reciprocal cause/effect relationship [2, 3]. On the other hand, degenerating conditions that affect joint function, such as osteoarthritis (OA) and inflammatory rheumatoid arthritis (RA), are the single largest cause of disability in the adult population [4, 5], and while our understanding of the pathology of these diseases has greatly improved, the only successful treatment for restoring joint function in end-stage diseases is total joint replacement [6]. To some extent, this lack of effective medical treatments is due to the fact that the articular cartilage is avascular, and this limits its reparative capacity [7]. Although physiologically and clinically significant, there is limited information on the mechanisms and signaling molecules that lead to joint morphogenesis. A greater understanding of joint formation can provide critical insight on the pathogenesis of joint degeneration and to develop novel reparative strategies to restore long-term joint function following trauma and disease.
Joint Morphogenesis
During skeletogenesis, long bone formation initiates as uninterrupted mesenchyme condensations in the early limb buds, which undergo differentiation to chondrocytes [1, 6]. Joint formation becomes morphologically evident with the appearance of a region called the interzone at the sites of future joint location where the resident cells flatten and form a clear separation of the previous uninterrupted cartilaginous skeletal anlagen [8, 9]. In most species, the interzone is a tripartite structure consisting of two outer layers adjacent to the epiphyseal end of the future bones and an intermediate zone that adopts a non-chondrogenic phenotype, as indicated by the loss of chondrogenic markers such as Sox-9 and Collagen 2 and the expression of new sets of genes including Gdf-5, Wnt-14, and Wnt-4 [10–12]. The pivotal role of the interzone in joint formation is widely recognized, as its microsurgical removal results in joint ablation [8, 9, 13]. Ultrastructural analysis in developing rat embryos indicated that the outer interzone layers participate in initial lengthening of long bone anlagen, while articular chondrocytes largely derive from the intermediate layer [14]. Despite wide recognition that the interzone region is essential for joint formation, there is limited information of the gene expression environment in which interzone cells emerge and distinguish themselves from adjacent growth plate chondrocytes [14–16]. Numerous studies in recent years have reported several signaling molecules, growth factors, transcription factors, and other regulatory molecules expressed by the interzone: these include GDF-5, Wnt-14, Wnt-4, Wnt-16, Gli3, and CD44; bone morphogenetic protein (BMP) antagonists Chordin and Noggin; fibroblast growth factor family members FGF-2, FGF-4, and FGF-13; and transcription factors Cux1 and ERG [10, 12, 17–20]. GDF-5 is expressed in the early interzone in mouse and chick embryo limb joints, and lack of GDF-5 in the natural mouse mutant brachypodism causes widespread joint defects and skeletal growth retardation [21]. Subsequent studies established that Gdf-5-expressing cells give rise to most if not all joint tissues, including articular cartilage, ligaments, and inner synovial lining [14]. The current view is that GDF-5 has at least two roles in skeletogenesis. At early stages, it is expressed throughout the condensation and would stimulate recruitment and differentiation of chondrogenic cells. At later stages, when its expression becomes restricted to the interzone, GDF-5 would promote interzone cell function and joint development [22]. Iwamoto et al. reported that there is a close spatiotemporal expression of GDF-5 with the transcriptional factor ERG (one of the ets gene family member) in developing mouse embryo joints, which suggests that ERG acts downstream of GDF-5 leading chondrocytes to become joint-forming cells [9].
Wnt-14 and the BMP antagonist Noggin are anti-chondrogenic factors that are expressed early during interzone formation. Targeted misexpression of Wnt-14 in developing chicken digit rays induced ectopic joints, with upregulation of Gdf-5, Wnt-4, and CD44 and downregulation of Col2 and Sox-9 [10], while ablation of Wnt-14 along with Wnt-4 impairs joint formation and causes some fusions [23]. In vitro studies show that Wnt-14 reversed chondrocyte differentiation in predifferentiated micromass culture, reflecting what happened in vivo [10, 24].
Experimental ablation of Noggin in mouse prevents limb joint formation and causes skeletal fusions [17]. The role of Noggin is conserved between mouse and humans, and at least two human syndromes, which are characterized by multiple synostoses (absence of joints), are due to mutations in Noggin [25]. The expression of another BMP antagonist, Chordin, is restricted to the joint interzone, emphasizing the role of BMP antagonism during joint development. The significant concept stemming from these studies is that joint formation involves the intervention of gene products with chondrogenic and anti-chondrogenic activities, the former critical for formation of fibrocartilaginous joint structures and the latter for maintaining the initial mesenchymal character of cells present at joint sites [15]. Thus, it is not surprising that the joint interzone appears to be also an essential regulator of skeletal development and vice versa [24]. One example is shown by the conditional inactivation of Ext1 in developing joints in Gdf-5-expressing cells. Ext1 encodes a subunit of the Ext1/Ext2 Golgi-associated protein complex responsible for heparan sulfate (HS) synthesis. HS is an essential component of cell surface- and matrix-associated proteoglycans, and mutations in HS-synthesizing and modifying enzymes cause several skeletal phenotypes, such as hereditary multiple exostoses (HME), described later in “Joint Genetic Diseases” section. Interestingly, mutation of Ext1 in Gdf-5-expressing cells led to lack of a distinct mesenchymal interzone with consequent joint fusion. They also found abnormal BMP and hedgehog activity and signaling leading to a delayed growth and lengthening of long bones, indicating that defects in joint formation reverberate on, and delay, overall long bone growth [26].
Recent studies have identified an important role for HIF-1 and HIF-2 in the regulation of skeletal development as well as joint formation and homeostasis. In addition, overexpression of HIF-1 and HIF-2 has been clinically associated with osteoarthritis [27].
The critical role of Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP) during endochondral bone formation is not only well known, but also their importance in synovial joint formation [28–30] has been reported. Ihh-null mice demonstrate failure in joint development [28, 31, 32] and have abnormal distribution and function of Gdf-5-expressing interzone-associated cells [17, 29, 31, 33]. These studies suggest that in the developing diaphysis, secreted Ihh could diffuse and reach the epiphyseal ends of the long bone anlagen, where it would regulate interzone formation [1]. Using a PTHrP-Lac-Z knockin mouse containing a β-Gal reporter under endogenous PTHrP gene regulatory sequences, Chen et al. have confirmed two distinct PTHrP β-Gal-positive subpopulations, in the articular cartilage and in the proliferative chondrocytes, that are maintained through adulthood [34] [35]. A role for PTHrP in articular cartilage maintenance has been recently proposed by Gdf-5-Cre-targeted knockdown of PTHrP in mouse articular chondrocytes that led to accelerated development of OA in a destabilization of the medial meniscus (DMM) model [36]. In addition, systemic administration of recombinant human PTH in mice inhibited cartilage degeneration and induced cartilage regeneration following meniscal/ligamentous injury [37].
Proper spatial positioning of synovial joints is crucial in skeletogenesis. Sohaskey et al. reported that mice carrying an insertional mutation in the Jaws gene (abnormal joint with splitting) die perinatally with striking skeletal defects, including ectopic interphalangeal joints [38]. These ectopic joints develop along the longitudinal axis and persist at birth, suggesting that JAWS is uniquely required for the orientation and consequent positioning of interphalangeal joints within the endochondral skeleton.
Transgenic mice overexpressing fibroblast growth factor receptors (FGFR) 1 and 3 exhibit joint defects similar to the symphalangism that occurs in Apert syndrome, a rare congenital disorder characterized by craniosynostosis, midfacial malformation, and symmetrical syndactyly. In these mice, some synovial joints failed to develop and the presumptive joint space was replaced by cartilage [39].
A recent study by Gao et al. reported a critical role of Osr1 and Osr2, the mammalian homologs of the odd-skipped family of zinc finger transcription factors required for Drosophila leg joint formation, in mouse synovial joint formation. They report that Osr1 and Osr2 are both strongly expressed in the developing synovial joint cells and are required for Gdf-5, Wnt-4, and Wnt-14 expressions [40].
Genetic manipulation of the TGF-β system has revealed critical roles in both joint development and skeletogenesis [41–43]. In transgenic mice, overexpression of a dominant-negative Tgfbr2 (DNIIR) resulted in OA [40]. In humans, mutations of the Tgfbr2 have been associated with Loeys-Dietz syndrome, a Marfan-like syndrome that leads to early onset of OA (OMIM #609192) [44, 45]. In previous studies, by generating a TGF-β type II receptor (TβRII) reporter mouse (β-Gal and GFP), we showed that TβRII is highly and specifically expressed in the developing joint [43]. By generating a mouse in which the TβRII signaling is conditionally inactivated in mesenchyme limb buds starting at E9.5 (Tgfbr2 Prx1KO), we demonstrated that the lack of TβRII signaling led to complete absence of interphalangeal joints and was associated with downregulation of key joint morphogenic factors (such as Noggin, GDF-5, and the Notch ligand Jagged 1). Interestingly, a lack of joint was associated with upregulation of many inflammatory cytokines/chemokines in the presumptive interzone, such as monocyte chemoattractant protein-5 (MCP-5) and interleukin 36 (IL-36) [43, 46, 47]; blockade of the MCP-5 receptor was able to rescue the TβRII-mediated joint deficiency [47], suggesting that cytokines/chemokines may have a regulatory role during development, mediating proper interzone formation.
There is compelling evidence that Notch signaling is critical for progenitor cell survival and for their maintenance in an undifferentiated state, which sets the boundaries that segregates two distinct cell populations during development [48–52]. Notch 1 is a regulator of great importance for cell fate commitment during both early growth and in adult tissues and has been shown to play a critical role in the cell fate determinant in different stem cell niche structures such as the skin, teeth, and nervous system [53]. Hayes et al. reported that Notch 1 is expressed in the developing and postnatal articular surface [54]. The Notch family members are also expressed in adult articular cartilage [55], and over 70 % of chondrocytes on the surface zone of articular cartilage express Notch 1 receptor [16], suggesting a significant role of the Notch signaling in regulating articular cartilage homeostasis during adult life [55, 56].
Despite important advances on multiple fronts, there are still fundamental aspects of joint formation that remain unclear and, in particular, how joints acquire their diverse morphology and organization from embryonic life to adulthood. A better understanding of genes involved in patterning processes will be critical to link embryonic development with joint disease in adult, in both inherited and acquired debilitating diseases.
Progenitor Stem Cell Niche
The view of articular cartilage as a non-regenerative organ has been challenged in recent years. The superficial zone of articular cartilage demonstrates a distinct pattern regarding stem cell markers (Notch 1, Stro-1, and vascular cell adhesion-1) [57] and has been hypothesized to harbor stem cells [58]. Studies regarding cartilage development have shed light on the question of whether any of the joint tissues harbor potential joint progenitor cells. Through the use of bromo-deoxyuridine (BrdU) injections to localize slow-cycling cells, a trait characteristic of progenitor cells, a few potential joint stem cell niches have been determined. A Notch 1-positive cell population has been isolated from the surface zone of articular cartilage [59]; these cells possess high colony-forming efficiency that was abolished when Notch signaling was blocked [16].
Karystinou et al. reported the isolation and characterization of multipotent mesenchymal stem cells (MSCs) from adult human synovium that could be expanded for up to 30 population doubling, with limited senescence and maintenance of multilineage differentiation capacity toward chondrogenesis, osteogenesis, adipogenesis, and skeletal myogenesis [60]. In response to injury of various types, including trauma, the synovial membrane rapidly becomes hyperplastic, which seems to be sustained mainly by stromal synovial fibroblasts [61–63]. The biologic function of synovial cell proliferation is believed to have a pivotal role in joint homeostasis, and deregulation of this process is thought to contribute to the formation of pannus, which in RA causes destruction of cartilage and bone [63]. Although very little is known about the identity of the synovial cells that proliferate following injury, this study provides the first evidence of the existence in vivo, within postnatal knee synovium, of non-hematopoietic, non-endothelial stromal cells with MSC phenotype [64–66] which proliferate following articular cartilage injury and, under specific experimental conditions, differentiate into chondrocytes in an area of cartilage metaplasia within the synovium [67].
An area of potential interest with regard to joint progenitor cells is the perichondrial groove of Ranvier. This area is located at the periphery of the epiphyseal growth plate and has been demonstrated to contain proliferating cells [68]. Studies by Karlsson et al. in the knee of sexually mature rabbits have proved the existence of different subpopulations of progenitor cells, in the articular cartilage, in the groove of Ranvier, as well as the epiphyseal plate near the groove of Ranvier; however, whereas the progenitor cells in the surface layer of the articular cartilage seem to lose their progenitor properties and become dispersed throughout the articular cartilage as they grow older, the progenitor cells in the groove of Ranvier were positive for several markers associated with stem cell niche (including Stro-1, Jagged 1, and BMPr1a) and maintained their progenitor properties and localization over time [58].
We have addressed in the previous section the critical role of the TβRII in joint element development. Our finding that mice lacking TβRII signaling in developing limbs demonstrated a failed interzone formation and lacked interphalangeal joints led us to investigate the expression pattern of TβRII-expressing cells and their characterization as joint progenitors [69]. By using a Tgfbr2-β-Gal-GFP-BAC mouse previously described, we have recently characterized the TβRII-expressing cells as joint progenitors [69], clustering in a contiguous niche that comprises the groove of Ranvier and the synovio-entheseal complex, including part of the perichondrium, the synovium, the articular cartilage superficial layer, and the tendon’s enthuses [69]. Developmental-stage studies showed that TβRII expression was in synchrony with expression of joint-morphogenic genes such as Noggin, Gdf-5, Notch 1, and Jagged 1. Prenatal and postnatal BrdU-incorporation studies showed that within this synovio-entheseal articular cartilage niche, most of the TβRII-expressing cells exhibit stem cell traits. In addition, TβRII-expressing cells isolated from embryonic limb mesenchyme expressed joint progenitor markers in a time- and TGF-β-dependent manner. TβRII-expressing cells were maintained in their specific niches from embryonic life throughout early adulthood, although characterized by a progressive age-dependent reduction in number.
The functional characterization of adult joint progenitors, together with the identification of embryonic markers that are preserved in postnatal joints, opens prospects for potential ways to reactivate the joint-forming ability of such cells, to implement their survival during aging, and to evaluate their role in joint genetic disease as well as joint degenerative pathologies, such as arthritis.
Joint Genetic Diseases
Genetic constitution is a factor in the development of most diseases, including those of bones and joints. Evidence for the influence of genes in most disease development is expressed in two ways. In polygenic disorders, the genetic influence is indirect, based only on the higher rate of recurrence in some families compared to the general population. The second group follows Mendelian inheritance, where a specific gene is directly responsible for the disease and the gene is passed on to subsequent generations. These diseases may also appear spontaneously as new mutations. There are over 5000 documented Mendelian disorders, and over 500 of these affect bones and joints [70]. Some of these single-gene disorders affect many tissues, and the skeletal system, including joints, is one of many organs involved [71]; others are directly related to joint malformation. It is beyond the scope of this review to give a detailed list of all the joint genetic diseases; however, we will provide some examples in Table 1.
There are very few genes that are known to be necessary and/or sufficient to initiate the joint formation process, among them Noggin, Gdf-5, Tgfbr2, and Wnt-14 [10, 17, 22, 41]. The factor(s) that induce the expression of these joint morphogenic molecules are undefined. Gong et al. [25] demonstrated that five dominant mutations in the Noggin gene in unrelated families segregate with proximal symphalangism (SYM1A, OMIM #185800), an autosomal dominant disorder characterized by a complete bony fusion between the proximal and middle phalanges of the digit. Another form of proximal symphalangism (SYM1B) is caused by a heterozygous missense mutation in the Gdf-5 (OMIM #615298).
We reported that TβRII functions as a master regulator for the expression of key joint marker genes, such as Gdf-5 and Noggin, and is necessary for proper joint development [43]. The joint phenotype observed in mice lacking the Tgfbr2 gene in limbs is similar to that in patients with SYM1. In humans, heterozygous mutations of the Tgfbr2 gene have been associated with Loeys-Dietz syndrome (OMIM #610168), a Marfan-like syndrome characterized by several defects including joint hypermotility and/or contractures that lead to early onset of OA [44, 45]. It has been hypothesized that in Loeys-Dietz syndrome, an excess of TGF-β signaling accounts for the pathological manifestations [72].
Several lines of evidence have demonstrated the importance of Notch signaling in joint morphogenesis and genetic inactivation of the Notch signaling in Drosophila, resulting in loss of joints [73]. A derangement of Notch signaling has been reported in articular synoviocytes from patients with RA, characterized by aberrant synoviocyte proliferation [74]. Heterozygous mutation in the Notch 2 gene causes the Hajdu-Cheney syndrome (HJCYS, OMIM #102500), a rare autosomal dominant disorder that, in addition to severe skeletal abnormalities including acroosteolysis, is characterized by joint laxity. Mutations and deletions in Jagged 1 (Notch ligand) have been found in patients with Alagille syndrome characterized by intrahepatic cholestasis and eye and cardiac abnormalities, as well as skeletal defects in particular in the fingers [75, 76].
Mouse studies have indicated that embryonic long bone development is altered by mutations in Ext gene members (Ext1, Ext2, and Ext3) of the exostosin protein family and responsible for the synthesis of heparan sulfate (HS) [77]. Recent data have shown that loss of Ext1 expression postnatally is not only affecting the growth and organization of long bones but is incompatible with normal joint structure, resembling human OA [78, 79]. In humans, HS deficiency due to mutation in the genes encoding exostosin-1, -2, and -3, causes multiple hereditary exostoses (EXT, OMIM #133700), an autosomal dominant disorder characterized by multiple projections of bone capped by cartilage, most numerous in the metaphyses of long bones, but also occurring on the diaphyses of long bones. Deformity of the legs, forearms (resembling Madelung deformity), and hands is frequent [80].
Mutations in the Jaws gene (joints abnormal with splitting), also known as inositol monophosphatase domain-containing protein 1 (Impad1), cause the GPAPP type of chondrodysplasia with joint contractures, an autosomal recessive disorder characterized by short stature, chondrodysplasia with brachydactyly, congenital joint dislocations, micrognathia, cleft palate, and facial dysmorphism (OMIM #614078).
Alkaptonuria (AKU, OMIM #203500) is an autosomal recessive metabolic disorder caused by homozygous or compound heterozygous mutations in the homogentisate 1,2-dioxygenase (HGD) gene and characterized by accumulation of homogentisic acid, leading to darkened urine; ochronotic pigmentation of fibrous tissues including cartilage, tendons, and ligaments; chronic joint pain; and destruction of the cardiac valves. Ultimately, patients develop severe secondary OA.
Current knowledge of joint biology and pathologies has not been translated, however, into effective therapies to treat joint conditions [103, 104]. Knowing how joints form during fetal life, express certain genes that become established in early postnatal life, and are maintained in adult life would shed light on the homeostasis of the whole joint and could lead to novel treatment for many joint disorders.
Can We Understand Better OA from Joint Development?
OA is a complex disease caused by the interaction of multiple genetic and environmental factors and the major cause of disability in the adult population. Although articular cartilage degeneration is the primary concern in OA, it is now the prevailing paradigm that OA is a disease of the entire joint and homeostasis and integrity of articular cartilage depend also on the biochemical interplay with subchondral bone; thus, it is not surprising that mutation in genes that are needed during skeletogenesis may lead to OA later in life.
We have addressed the critical role of the TGF-β system in both joint development and skeletogenesis [41–43]. The scope of this section is to provide an example of how genes that are so critical in joint morphogenesis may also have key roles in joint-degenerating disease, such as OA. TGF-βs bind to TβRII leading to TβRII-TβRI complex formation, which then activates the signaling cascade through R-Smad-dependent (Smad-2, -3, -4) and Smad-independent pathways [81, 82].
In human and mouse embryonic cartilage, TGF-βs are expressed in the endochondral template [83–86]. Using laser capturing microdissection (LCM), we have obtained RNA samples from E13.5 progenitor joint-forming interzone cells and from the adjacent growth plate chondrocytes. Each population-related mRNA was subjected to microarray analysis. We have found that Tgfbr2 expression was 12.3 times higher, while Tgfbr1 and Tgfbr3 were slightly lower in the interzone cells. In mice, targeted manipulation of the TGF-β system genes has revealed their critical, but still undefined, function in skeletogenesis [41, 42, 87, 88]. R-Smad gene targeting in mice has led to variable phenotypes ranging from early lethality (Smad-2 and Smad-4 ablation) to a normal phenotype at birth with progressive OA in adulthood (Smad-3 ablation) [89–92]. In transgenic mice, overexpression of a dominant-negative Tgfbr2 (DNIIR) can result in OA in adults [42]; in chick embryos, however, implanted beads releasing TGF-β induce extra digit formation [93]. In humans, mutations of the Tgfbr2 have been associated with Loeys-Dietz syndrome [44, 45, 72].
In previous studies, we demonstrated that lack of TβRII signaling in developing limbs was associated with complete absence of interphalangeal joints as well as with impaired growth plate development [43].
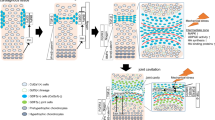
By combining LCM with microarray analysis, we were also able to show that interzone cells express a gene pattern that is distinct from adjacent growth plate chondrocytes and such gene regulation is impaired in mutant limbs lacking TβRII. We performed a pathway analysis using the Protein Analysis Through Evolutionary Relationships (PANTHER) Classification System (Celera) to classify genes into canonical pathways. This enabled us to identify functional categories in the joint-forming interzone cells of wild type mice that were different from mutants. We determined that in the interzone cells, the “inflammation mediated by chemokines and cytokines” was the most downregulated pathway in the wild type interzone compared to that in the mutant. Specifically, we found that MCP-5 and IL-36α were among the most downregulated genes in this pathway [46, 47]. It has been reported that MCP-5, MCP-1, and their sole common receptor CCR2, as well as IL-36α, are increased in the inflamed joints of patients with different forms of arthritis and in rodent models for arthritis [94–97]. While the role of inflammatory cytokines in the arthritic process appears indisputable, their role in joint development has never been evaluated. A finding of cytokine involvement in joint formation would determine a shift in our current view of joint cells not only as passive victims of the disruptive force of cytokines but as active participants in maintaining control of the cytokine effects and therefore joint integrity. It has been shown that in healthy cartilage, a balance between cytokines and anabolic growth factors is critical to maintain proper tissue homeostasis [98, 99]. Supported by our previously published data and other current evidence, in Fig. 1, we propose a model for the role of TβRII signaling in regulating cell commitment to joint-forming cells or chondrocytes within the joints [47]. Specifically, TβRII signaling, by blocking critical inflammatory cytokine expression in the interzone, halts Collagen 2 expression and induces interzone-specific markers (i.e., Gdf-5, Noggin). In mutant mice, lack of the TβRII expression in developing limbs leads to dysregulated levels of cytokines/chemokines in the interzone (such as MCP-5 and IL-36α which, in turn, block expression of interzone-specific markers while increasing Collagen 2-expressing cells), with consequent accumulation of chondrocytes within the joint region and impaired joint formation.

A model for the TβRII-mediated regulation of joint development. TβRII downregulates MCP-5/IL-36α expressions in the interzone, allowing expression of key joint markers (such as Gdf-5) while inhibiting Collagen 2 expression. Lack of the TβRII signaling leads to upregulation of interzone MCP-5/IL-36α with consequent lack of joint formation
The activation of the inflammatory cytokine/chemokine cascade is associated with the more common forms of arthritis [100]. The generally accepted hypothesis is that during the OA/RA process, the increased levels of cytokines and chemokines are the last step of a long-lasting damage process that sees the joint as a passive target. To determine the role of TβRII signaling in joint homeostasis, we generate a genetically modified mouse to tamoxifen which induces a Cre-conditional inactivation of floxed Tbfbr2 in osteochondro-progenitors starting at postnatal day 3 of life [46]. We found that postnatal TβRII signaling inactivation was associated with high expression of IL-36α and MCP-5 (unpublished data) in the articular cartilage and synovium and progressive OA development [46]. Blockage of IL-36α signaling led to rescue of the OA phenotype [46]. In a mouse model of posttraumatic OA, namely caused by a DMM surgically induced after medial meniscotibial ligament transection, we found an intense early cellular response of TβRII-positive cells that gradually decreased with OA progression and was associated with an increase of MCP-5 [101]. Blockage of the MCP-5 signaling as well as implant of TβRII-positive cells improved OA lesions and decreased subchondral bone reaction [101, 102]. These findings suggest that TβRII signaling and cellular response are critical in joint development and these are active mechanisms to preserve joint homeostasis in adulthood. In this respect, we provide a novel perspective to evaluate the OA process that results from the failure of an active cell joint population, expressing TβRII, to maintain a controlled cytokine environment rather than from the damage induced by a systemic influx of inflammatory cells and cytokines against a passive target (the joint).
Conclusions
Joint biology has been the subject of extensive research activity, and much has been learned about the structure and composition of articular cartilage, ligaments, synovium, and joint capsule and the specific roles each of these tissues plays in joint function in developing and adult organisms. Much is also known about susceptibility of joint tissues to damage and malfunction during natural aging and congenital or acquired skeletal conditions [20]. Diseases that target and disrupt structure and function in the articular cartilage, synovium, meniscus, and subchondral bone result in painful, debilitating, and costly conditions for patients and society as a whole. The articular cartilage is avascular, and this presents limitations on its reparative capacity and for drug delivery. Joint homeostasis is maintained, preserving the articular cells to proceed down the differentiation pathway that leads to chondrocyte hypertrophy and bone replacement such as it occurs in the growth plate cartilage. Many joint-degenerating conditions may be seen as the failure of morphogenic factors to establish the correct microenvironment in cartilage and bone. Therefore, exploring how joints form can lead us to understand how cartilage and bone are damaged and to develop drugs that are able to reactivate this developing mechanism. This information would have major biomedical relevance, as it could lead to novel repair strategies to restore joint function following trauma, as well as provide insight on the pathogenesis of acquired and inherited joint diseases.
References
Pacifici M, Koyama E, Iwamoto M. Mechanisms of synovial joint and articular cartilage formation: recent advances, but many lingering mysteries. Birth Defects Res C Embryo Today. 2005;75(3):237–48 [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S. Review].
Ikegawa S. Genetic analysis of skeletal dysplasia: recent advances and perspectives in the post-genome-sequence era. J Hum Genet. 2006;51(7):581–6.
Pauli RM. The natural histories of bone dysplasias in adults—vignettes, fables and just-so stories. Am J Med Genet C: Semin Med Genet. 2007;145C(3):309–21.
Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213(3):626–34.
Hunter DJ. In the clinic. Osteoarthritis. Ann Intern Med. 2007;147(3):ITC8. -1-ITC8-16.
Khan IM, Redman SN, Williams R, Dowthwaite GP, Oldfield SF, Archer CW. The development of synovial joints. Curr Top Dev Biol. 2007;79:1–36 [Review].
Redman SN, Oldfield SF, Archer CW. Current strategies for articular cartilage repair. Eur Cell Mater. 2005;9:23–32. discussion 23–32.
Hyde G, Dover S, Aszodi A, Wallis GA, Boot-Handford RP. Lineage tracing using matrilin-1 gene expression reveals that articular chondrocytes exist as the joint interzone forms. Dev Biol. 2007;304(2):825–33.
Iwamoto M, Tamamura Y, Koyama E, Komori T, Takeshita N, Williams JA, et al. Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev Biol. 2007;305(1):40–51.
Hartmann C, Tabin CJ. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell. 2001;104(3):341–51.
Pacifici M, Koyama E, Shibukawa Y, Wu C, Tamamura Y, Enomoto-Iwamoto M, et al. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Ann N Y Acad Sci. 2006;1068:74–86 [Research Support, N.I.H., Extramural Review].
Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994;368(6472):639–43.
Holder N. An experimental investigation into the early development of the chick elbow joint. J Embryol Exp Morpholog. 1977;39:115–27.
Koyama E, Shibukawa Y, Nagayama M, Sugito H, Young B, Yuasa T, et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev Biol. 2008;316(1):62–73.
Koyama E, Ochiai T, Rountree RB, Kingsley DM, Enomoto-Iwamoto M, Iwamoto M, et al. Synovial joint formation during mouse limb skeletogenesis: roles of Indian hedgehog signaling. Ann N Y Acad Sci. 2007;1116:100–12.
Dowthwaite GP, Bishop JC, Redman SN, Khan IM, Rooney P, Evans DJ, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci. 2004;117(Pt 6):889–97.
Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280(5368):1455–7.
Iwamoto M, Higuchi Y, Koyama E, Enomoto-Iwamoto M, Kurisu K, Yeh H, et al. Transcription factor ERG variants and functional diversification of chondrocytes during limb long bone development. J Cell Biol. 2000;150(1):27–40.
Lizarraga G, Lichtler A, Upholt WB, Kosher RA. Studies on the role of Cux1 in regulation of the onset of joint formation in the developing limb. Dev Biol. 2002;243(1):44–54.
Archer CW, Dowthwaite GP, Francis-West P. Development of synovial joints. Birth Defects Res C Embryo Today. 2003;69(2):144–55.
Settle Jr SH, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003;254(1):116–30.
Storm EE, Kingsley DM. GDF5 coordinates bone and joint formation during digit development. Dev Biol. 1999;209(1):11–27.
Spater D, Hill TP, O’Sullivan RJ, Gruber M, Conner DA, Hartmann C. Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development. 2006;133(15):3039–49.
Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang T. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18(19):2404–17.
Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R, et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet. 1999;21(3):302–4.
Mundy C, Yasuda T, Kinumatsu T, Yamaguchi Y, Iwamoto M, Enomoto-Iwamoto M, et al. Synovial joint formation requires local Ext1 expression and heparan sulfate production in developing mouse embryo limbs and spine. Dev Biol. 2011;351(1):70–81.
Husa M, Liu-Bryan R, Terkeltaub R. Shifting HIFs in osteoarthritis. Nat Med. 2010;16(6):641–4. doi:10.1038/nm0610-641.
Rankin EB, Giaccia AJ, Schipani E. A central role for hypoxic signaling in cartilage, bone, and hematopoiesis. Curr Osteoporos Rep. Jun;9(2):46–52.
Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273(5275):663–6 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613–22.
Wallis GA. Bone growth: coordinating chondrocyte differentiation. Curr Biol. 1996;6(12):1577–80 [Review].
Koyama E, Leatherman JL, Noji S, Pacifici M. Early chick limb cartilaginous elements possess polarizing activity and express hedgehog-related morphogenetic factors. Dev Dyn. 1996;207(3):344–54.
Kohno H, Shigeno C, Kasai R, Akiyama H, Iida H, Tsuboyama T, et al. Synovial fluids from patients with osteoarthritis and rheumatoid arthritis contain high levels of parathyroid hormone-related peptide. J Bone Miner Res. 1997;12(5):847–54 [Clinical Trial Controlled Clinical Trial Research Support, Non-U.S. Gov’t].
Nakamura T, Aikawa T, Iwamoto-Enomoto M, Iwamoto M, Higuchi Y, Pacifici M, et al. Induction of osteogenic differentiation by hedgehog proteins. Biochem Biophys Res Commun. 1997;237(2):465–9.
Mountford P, Zevnik B, Duwel A, Nichols J, Li M, Dani C, et al. Dicistronic targeting constructs: reporters and modifiers of mammalian gene expression. Proc Natl Acad Sci U S A. 1994;91(10):4303–7 [Research Support, Non-U.S. Gov’t].
Chen X, Macica CM, Dreyer BE, Hammond VE, Hens JR, Philbrick WM, et al. Initial characterization of PTH-related protein gene-driven lacZ expression in the mouse. J Bone Miner Res. 2006;21(1):113–23 [Research Support, N.I.H., Extramural].
Sampson ER, Hilton MJ, Tian Y, Chen D, Schwarz EM, Mooney RA, et al. Teriparatide as a chondroregenerative therapy for injury-induced osteoarthritis. Sci Transl Med. 2011;3(101):101ra93 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Sohaskey ML, Yu J, Diaz MA, Plaas AH, Harland RM. JAWS coordinates chondrogenesis and synovial joint positioning. Development. 2008;135(13):2215–20.
Wang Q, Green RP, Zhao G, Ornitz DM. Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development. 2001;128(19):3867–76.
Gao Y, Lan Y, Liu H, Jiang R. The zinc finger transcription factors Osr1 and Osr2 control synovial joint formation. Dev Biol. 2011;352(1):83–91.
Ito Y, Yeo JY, Chytil A, Han J, Bringas Jr P, Nakajima A, et al. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130(21):5269–80.
Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, et al. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139(2):541–52.
Spagnoli A, O’Rear L, Chandler RL, Granero-Molto F, Mortlock DP, Gorska AE, et al. TGF-beta signaling is essential for joint morphogenesis. J Cell Biol. 2007;177(6):1105–17.
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–81.
Jones KB, Sponseller PD, Erkula G, Sakai L, Ramirez F, Dietz 3rd HC, et al. Symposium on the musculoskeletal aspects of Marfan syndrome: meeting report and state of the science. J Orthop Res. 2007;25(3):413–22.
Li T, Longobardi L, Myers TJ, Temple JD, Esposito A and Spagnoli A. Tgfbeta signaling regulates interleukin-36 alpha in joint development and osteoarthritis. Abstract presented to ASBMR meeting. 2013.
Longobardi LLT, Myers TJ, O’Rear L, Ozkan H, Li Y, Contaldo C and Spagnoli A. TGF-β type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Dev Cell. 2012.
Hansson EM, Lendahl U, Chapman G. Notch signaling in development and disease. Semin Cancer Biol. 2004;14(5):320–8.
Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131(5):965–73.
Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev. 2006;7(9):678–89.
Milner LA, Bigas A. Notch as a mediator of cell fate determination in hematopoiesis: evidence and speculation. Blood. 1999;93(8):2431–48.
Chiba S. Notch signaling in stem cell systems. Stem Cells Dayton Ohio. 2006;24(11):2437–47.
Lovschall H, Mitsiadis TA, Poulsen K, Jensen KH, Kjeldsen AL. Coexpression of Notch3 and Rgs5 in the pericyte-vascular smooth muscle cell axis in response to pulp injury. Int J Dev Biol. 2007;51(8):715–21.
Hayes AJ, Dowthwaite GP, Webster SV, Archer CW. The distribution of Notch receptors and their ligands during articular cartilage development. J Anat. 2003;202(6):495–502.
Karlsson C, Brantsing C, Egell S, Lindahl A. Notch1, Jagged1, and HES5 are abundantly expressed in osteoarthritis. Cells Tissues Organs. 2008;188(3):287–98.
Sassi N et al. The role of the Notch pathway in healthy and osteoarthritic articular cartilage: from experimental models to ex vivo studies. Arthritis Res Ther. 2011;13(2):208.
Grogan SP, Miyaki S, Asahara H, D’Lima DD, Lotz MK. Mesenchymal progenitor cell markers in human articular cartilage: normal distribution and changes in osteoarthritis. Arthritis Res Ther. 2009;11(3):R85.
Henriksson H, Thornemo M, Karlsson C, Hagg O, Junevik K, Lindahl A, et al. Identification of cell proliferation zones, progenitor cells and a potential stem cell niche in the intervertebral disc region: a study in four species. Spine (Phila Pa 1976). 2009;34(21):2278–87.
Hayes AJ, Benjamin M, Ralphs JR. Extracellular matrix in development of the intervertebral disc. Matrix Biol. 2001;20(2):107–21.
Karystinou A, Dell’Accio F, Kurth TB, Wackerhage H, Khan IM, Archer CW, et al. Distinct mesenchymal progenitor cell subsets in the adult human synovium. Rheumatology (Oxford). 2009;48(9):1057–64.
Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126(6):1611–23 [Research Support, Non-U.S. Gov’t].
Pessler F, Dai L, Diaz-Torne C, Gomez-Vaquero C, Paessler ME, Zheng DH, et al. The synovitis of “non-inflammatory” orthopaedic arthropathies: a quantitative histological and immunohistochemical analysis. Ann Rheum Dis. 2008;67(8):1184–7.
Buckley CD, Filer A, Haworth O, Parsonage G, Salmon M. Defining a role for fibroblasts in the persistence of chronic inflammatory joint disease. Ann Rheum Dis. 2004;63 Suppl 2:ii92–ii5.
De Bari C, Dell’Accio F, Luyten FP. Human periosteum-derived cells maintain phenotypic stability and chondrogenic potential throughout expansion regardless of donor age. Arthritis Rheum. 2001;44(1):85–95 [Research Support, Non-U.S. Gov’t].
De Bari C, Dell’Accio F, Vandenabeele F, Vermeesch JR, Raymackers JM, Luyten F, et al. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J Cell Biol. 2003;160(6):909–18 [Research Support, Non-U.S. Gov’t].
De Bari C, Dell’Accio F, Luyten FP. Failure of in vitro-differentiated mesenchymal stem cells from the synovial membrane to form ectopic stable cartilage in vivo. Arthritis Rheum. 2004;50(1):142–50 [In Vitro Research Support, Non-U.S. Gov’t].
De Bari C, Kurth TB, Augello A. Mesenchymal stem cells from development to postnatal joint homeostasis, aging, and disease. Birth Defects Res C Embryol Today. 2010;90(4):257–71 [Research Support, Non-U.S. Gov’t Review].
Shapiro F, Holtrop ME, Glimcher MJ. Organization and cellular biology of the perichondrial ossification groove of ranvier: a morphological study in rabbits. J Bone Joint Surg Am. 1977;59(6):703–23.
Li T, Longobardi L, Myers TJ, Temple JD, Chandler RL, Ozkan H, et al. Joint TGF-beta type II receptor-expressing cells: ontogeny and characterization as joint progenitors. Stem Cells Dev. May 1;22(9):1342–59.
McKusick VA. First South-North Human Genome Conference. Genomics. 1992;14(4):1121–3.
McCarthy EF. Genetic diseases of bones and joints. Semin Diagn Pathol. Feb;28(1):26–36.
Ramirez F, Dietz HC. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17(3):252–8.
Shirai T, Yorimitsu T, Kiritooshi N, Matsuzaki F, Nakagoshi H. Notch signaling relieves the joint-suppressive activity of defective proventriculus in the Drosophila leg. Dev Biol. 2007;312(1):147–56.
Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T, et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene. 2003;22(49):7796–803.
Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235–42.
Boyer J, Crosnier C, Driancourt C, Raynaud N, Gonzales M, Hadchouel M, et al. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet. 2005;116(6):445–53.
Busse M, Feta A, Presto J, Wilen M, Gronning M, Kjellen L, et al. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem. 2007;282(45):32802–10.
Mundy C, Yasuda T, Kinumatsu T, Yamaguchi Y, Iwamoto M, Enomoto-Iwamoto M, et al. Synovial joint formation requires local Ext1 expression and heparan sulfate production in developing mouse embryo limbs and spine. Dev Biol. Mar 1;351(1):70–81.
Sgariglia F, Candela ME, Huegel J, Jacenko O, Koyama E, Yamaguchi Y, et al. Epiphyseal abnormalities, trabecular bone loss and articular chondrocyte hypertrophy develop in the long bones of postnatal Ext1-deficient mice. Bone. Nov;57(1):220–31.
Peterson HA. Multiple hereditary osteochondromata. Clin Orthop. 1989;239:222–30.
Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580(12):2811–20.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84.
Lawler S, Candia AF, Ebner R, Shum L, Lopez AR, Moses HL, et al. The murine type II TGF-beta receptor has a coincident embryonic expression and binding preference for TGF-beta 1. Development. 1994;120(1):165–75.
Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115(4):1091–105.
Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111(1):131–43.
Serra R, Chang C. TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res C Embryo Today. 2003;69(4):333–51 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review].
Dunker N, Krieglstein K. Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur J Biochem. 2000;267(24):6982–8.
Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276(1):124–42.
Weinstein M, Yang X, Li C, Xu X, Gotay J, Deng CX. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci U S A. 1998;95(16):9378–83.
Yang X, Chen L, Xu X, Li C, Huang C, Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153(1):35–46.
Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94(6):703–14.
Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12(1):107–19.
Ganan Y, Macias D, Duterque-Coquillaud M, Ros MA, Hurle JM. Role of TGF betas and BMPs as signals controlling the position of the digits and the areas of interdigital cell death in the developing chick limb autopod. Development. 1996;122(8):2349–57.
Robinson E, Keystone EC, Schall TJ, Gillett N, Fish EN. Chemokine expression in rheumatoid arthritis (RA): evidence of RANTES and macrophage inflammatory protein (MIP)-1 beta production by synovial T cells. Clin Exp Immunol. 1995;101(3):398–407.
Hayashida K, Nanki T, Girschick H, Yavuz S, Ochi T, Lipsky PE. Synovial stromal cells from rheumatoid arthritis patients attract monocytes by producing MCP-1 and IL-8. Arthritis Res. 2001;3(2):118–26.
Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90(3):772–9.
De Benedetti F, Pignatti P, Bernasconi S, Gerloni V, Matsushima K, Caporali R, et al. Interleukin 8 and monocyte chemoattractant protein-1 in patients with juvenile rheumatoid arthritis. Relation to onset types, disease activity, and synovial fluid leukocytes. J Rheumatol. 1999;26(2):425–31.
Pujol JP, Chadjichristos C, Legendre F, Bauge C, Beauchef G, Andriamanalijaona R, et al. Interleukin-1 and transforming growth factor-beta 1 as crucial factors in osteoarthritic cartilage metabolism. Connect Tissue Res. 2008;49(3):293–7.
Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol. 2008;40(10):2174–82.
Szekanecz Z, Halloran MM, Volin MV, Woods JM, Strieter RM, Kenneth Haines 3rd G, et al. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43(6):1266–77.
Lara Longobardi JDT, nunzia D’Onofrio, Ozkan H, Esposito A, Willcockson HH, Li T, Myers TJ, Ye P, Moats-Staats BM, Balestrieri M and Spagnoli A. A role for Tgf-betaRII/MCP5 axis during post-traumatic osteoarthritis and potential role od PTHrP in mediating MCP5 effect. Abstract presented at the OARSI meeting, Paris 2014.
Lara Longobardi HO, Temple JD, Li T, Esposito A, Myers TJ, and Spagnoli A. Inhibition of MCP5 signaling decreases osteoarthritis lesions in a murine model of post-traumatic osteoarthritis. Abstract presented at the ASBMR Meeting, Baltimore, MD, USA. 2013.
Bird HA. Controversies in the treatment of osteoarthritis. Clin Rheumatol. 2003;22(3):165–7.
Brandt KD. Non-surgical treatment of osteoarthritis: a half century of “advances”. Ann Rheum Dis. 2004;63(2):117–22.
Acknowledgments
This work was supported by a NIH-NIAMS Grant 1R01AR057042-03 (to AS), NC-TraCS Institute (NIH-CTSA) (to LL), NIH-NIAMS Grant 1R03AR063232-01 (to LL), and Arthritis National Research Foundation (to LL).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
L Longobardi, T Li, L Tagliafierro, JD Temple, HH Willcockson, P Ye, A Esposito, F Xu, and A Spagnoli all declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Skeletal Development
Rights and permissions
About this article

Cite this article
Longobardi, L., Li, T., Tagliafierro, L. et al. Synovial Joints: from Development to Homeostasis. Curr Osteoporos Rep 13, 41–51 (2015). https://doi.org/10.1007/s11914-014-0247-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-014-0247-7