
Abstract
Purpose of Review
Oncolytic viruses (OVs) exert their antitumor effect through selective killing of cancer cells and induction of host anti-tumor immunity. This review aims to summarize the recent and current trials with OVs for the treatment of lung cancer.
Recent Findings
Several OVs have been developed for the treatment of lung cancer including adenovirus, coxsackievirus B3, reovirus, and vaccinia virus and trials have demonstrated a safe toxicity profile. Early-phase trials in lung cancer with OVs have reported antiviral immune responses and evidence of clinical benefit. However, clinical efficacy of OVs in lung cancer either as monotherapy or in combination with chemotherapy has not been confirmed in larger phase II or III trials. Development of OVs in lung cancer has been limited by difficulty in administering OVs in the tumor directly as well as achieving adequate viral load at all tumor sites with systemically administered OVs.
Summary
Developing novel combinations with OVs, especially checkpoint inhibitors and other immunotherapeutics, may be a strategy to address the limited success seen thus far. Integrating appropriate biomarker studies and meaningful endpoints in future clinical trials will be imperative. Using novel viral delivery systems in addition to increasing tumor specificity through improved genetic modifications in the OVs are other strategies to improve efficacy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
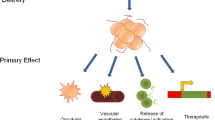
Oncolytic viruses (OVs) are a class of cancer therapeutics that exert their antitumor activity through selective killing of cancer cells and induction of host anti-tumor immunity [1•]. OVs are naturally occurring or genetically modified viruses that are engineered to replicate selectively in tumor cells leading to cell lysis while sparing normal host cells [2]. They can also modify the immunosuppressive microenvironment by altering the cytokine milieu and immune cells in the tumor microenvironment [3, 4]. The first OV approved in 2004 was a ribonucleic acid (RNA) virus derived from the native ECHO-7 strain of picornavirus, called Rigvir, for melanoma treatment in Latvia [5]. Other approvals since then include Oncorine (H101; adenovirus serotype 5) in China for head and neck cancer (2005), and Delytact (teserpaturev; modified Herpes simplex virus) in Japan for brain tumors (2021) [6, 7]. The only OV approved in the USA is talimogene laherparepvec (T‐VEC) for the local treatment of unresectable melanoma recurrent after initial surgery (2015) [1•, 8•]. T-VEC is a genetically modified herpes simplex type 1 (HSV-1) virus that contains a gene insertion encoding GM-CSF which promotes recruitment and activation of antigen presenting cells (APCs) [1•, 8•]. With T-VEC, despite the intratumoral injection, even the uninjected lesions and visceral metastases showed regression, likely due to the immune response elicited by the virus, implying that the induction of local antitumor immunity can have systemic effects [8•]. While currently none of the OVs have been approved for lung cancer, several have been tested in preclinical studies and clinical trials such as adenovirus, coxsackievirus B3, herpes virus, reovirus, and vaccinia virus [9•].
Genetic manipulation of the viral genome is the main strategy for OV development with the goals of weakening virus pathogenicity, enhancing target selectivity, reducing adverse reactions, and/or inserting exogenous therapeutic genes into the virus genome [10]. Using attenuated vectors or naturally occurring less virulent variants of viruses prevents acute infection as well as latent or chronic disease [1•]. Therefore, severe adverse events with OVs are rare and treatment discontinuation rate due to toxicity is low [11]. The majority of common treatment-related adverse events from OVs were low grade constitutional symptoms and local injections site reactions (12). For example, deletion of ICP6 in the HSV-1 genome weakens the pathogenicity of the virus and deletion of the gamma 34.5 gene enhances the neurotoxicity of HSV-1 and its selective replication in tumor cells [10]. To develop T-VEC, the ICP-34.5 gene in HSV1716 is deleted, which is a neurovirulence factor of HSV, attenuating the infectivity of HSV in normal neurons [8•, 13]. Mutation and deletion of the E1 gene can reduce adenovirus selectivity of normal cells, and this genetic modification was made to the Onyx-015 and H10 viruses [14, 15]. Therefore, normal cells infected with these viruses undergo p53-mediated abortive apoptosis, whereas tumor cells with inactivated p53 remain susceptible to viral lysis.
A recent review of 97 published OV trials reported that most OVs tested have used large deoxyribonucleic acid (DNA) viruses such as adenovirus (31%), HSV-1 (24%), reovirus (20%), and poxviruses (12%) [12]. Approximately two-thirds of trials used modified or recombinant viral backbones. GM-CSF, a cytokine that promotes recruitment and maturation of dendritic cells, was the most common transgene used. DNA viruses have a number of advantages including a large genome that can be edited without impairing viral replication. RNA viruses are smaller than DNA viruses but can cross the blood–brain barrier, enabling targeting of tumors in the central nervous system [16]. Intratumoral injection is the preferred OV delivery choice due to safety concerns after intravenous injection, or, to minimize the chance that preexisting circulating antibodies might neutralize the virus before it reaches its target [17]. However, direct intratumoral injection is a challenge for thoracic cancers due to the presence of visceral and/or multisite disease. Targeting thoracic tumors using interventional radiology or minimally invasive surgical approaches is being explored [9•].
Oncolytic Viruses and Tumor Immune Response
OVs modify the immunosuppressive microenvironment by altering the cytokine milieu and the immune cells in the tumor microenvironment [3, 4]. OV-mediated cell killing is associated with the release of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular pattern signals (DAMPs) such as heat shock proteins, ATP, and uric acid that are recognized by innate immune cells [1•, 18]. PAMPs and DAMPs also mediate the release of cytokines including IFNs, TNFα, IFNγ, and IL-12, which promote the maturation of APCs such as dendritic cells. These in turn activate antigen-specific CD4+ and CD8+ T cell responses [1•]. IFN released in response to OVs may also upregulate PD-L1 expression in tumor cells [19]. These changes promote immune-mediated tumor cell recognition and eradication and can trigger tumor-associated antigen and epitope spreading [20, 21]. In a preclinical model of CMT64 lung adenocarcinoma cells refractory to PD1 blockade, treatment with an oncolytic adenovirus was associated with oligoclonal neoantigen CD8+ T cell responses, whereas PD1 blockade induced only a monoclonal response [22]. On the other hand, neutralizing antibodies from prior viral exposures may block virus replication and ongoing infection of tumor cells. The therapeutic outcome, therefore, depends on a complex interplay between these opposing processes [20, 21].
OVs are still not powerful enough, especially for low immunogenic or immunosuppressive solid tumors [23]. A recent review of 97 published OV trials reported that the average objective response across the studies was 9% with an additional 12% of patients achieving stable disease as the best response [12]. Considerable work is being done to optimize viral vectors by genetic engineering to enhance immunogenicity [24]. Some viruses, such as reovirus, have inherent selectivity to tumor cells [25]. On the other hand, other OVs, such as adenovirus and HSV-1, have been genetically engineered to function as vectors to boost anti-tumor immune responses [26]. One common modification is the addition of IFNβ [27, 28]. IFNβ is the key innate mechanism to inhibit viral replication in healthy human cells. Gene therapy utilizing a replication-deficient adenovirus engineered to express IFNβ was tested in a phase I clinical trial for patients with lung cancer and mesothelioma with malignant pleural effusion. The viral vector was administered through direct instillation into the pleural space. Four of 10 patients experienced stable disease, but no responses were observed. Evidence of stimulation of antitumor immune responses was observed in seven of 10 patients [29•].
The combination of OVs with checkpoint inhibitors is thought to be synergistic by creating and then sustaining an immunogenic, anti-tumor microenvironment [30]. Combination of oncolytic adenoviruses with anti-PD-L1 and anti-CTLA-4 in preclinical studies showed the combination synergistically enhanced the antitumor effect by recruiting CD8+ T and T memory cells, reducing the number of regulatory T cells and tumor-associated macrophages, and promoting the polarization of macrophages from the M2 to the M1 phenotype [31]. In a phase Ib trial of patients with advanced melanoma, 21 patients were treated with T-VEC followed by combination therapy with pembrolizumab [32]. This combination therapy was well tolerated and showed no dose-limiting toxicities. The objective response rate (ORR) and complete response rate were 62% and 33%, respectively. Patients who responded to combination therapy demonstrated increased intratumoral CD8+ T cells, elevated PD-L1 expression, and IFN-γ gene expression after T-VEC treatment.
Oncolytic Viruses in Development for Lung Cancer
Oncolytic virus trials completed for the treatment of lung cancer have been listed in Table 1 and ongoing trials are listed in Table 2.
Coxsackeivirus
Coxsackievirus, a non-enveloped single-stranded RNA enterovirus, is a member of the Picornaviridae family [1•]. Coxsackievirus replicates in the cytoplasm without a DNA phase, eliminating the possibility of insertional mutagenesis during infection. It also does not require complex genetic manipulation for safety or oncolytic activity [1•]. Coxsackievirus A21 (Cavatak; CVA21) has a natural tropism for cancer cells as some tumors such as multiple myeloma, melanoma, and breast cancer, overexpress ICAM-1 and/or DAF for cell entry [33]. In addition to direct lysis of tumor cells, coxsackievirus enhances the immune response by promoting the release of DAMPs such as HMGB-1, calreticulin, and ATP [34]. A key factor to support use of coxsackievirus is its efficacy for pretreated non-small cell lung cancer (NSCLC). An appreciable antitumor effect has been reported with intratumoral coxsackievirus B3 (CVB3) administration against A549 lung adenocarcinoma xenografts, which were resistant to radiation treatment and EGFR tyrosine kinase inhibitor gefitinib [35, 36]. The overexpression of DAF by NSCLC cells provides a protective mechanism to counterbalance complement-mediated cytotoxicity and at the same time provide an excellent target for CVB3. Preclinical studies also report CVB3 induced caspase-mediated apoptosis and subsequent oncolysis in human NSCLC cells [34, 37].
Results from the phase 1b STORM trial (NCT02043665; KEYNOTE-200) were reported at the annual American Association for Cancer Research (AACR) meeting in 2017 [38]. Patients received CVA21 in escalating doses as monotherapy or in combination therapy with pembrolizumab across various advanced solid tumors. CVA21 was administered on days 1, 3, 5, 22, and every three weeks for six additional infusions. Pembrolizumab was given to patients for up to two years. CVA21 tumor targeting in patients was confirmed by detection of CVA21 viral RNA in tumor biopsies. All patients displayed active host-antiviral immune responses by developing detectable anti-CVA21 neutralizing antibodies by study Day 22. Of the 13 patients eligible for response assessment, one partial response and four stable disease were observed. The treatment was generally well-tolerated with at present no dose-limiting toxicities (DLTs). In the NSCLC expansion cohort [39], ORR in evaluable IO-naïve NSCLC patients to date is 23% (7/31, 2CR + 5PR) and 33% (7/21) in patients without EGFR or ALK mutations. Preliminary immunohistochemistry (IHC) staining of paired biopsies from evaluable patients with negative or low baseline PD-L1 expression revealed an increase in PD-L1 + tumor cells at Day 15 of 62% following combination treatment. While efficacy data is still immature at this time, the current median overall survival (OS) for patients with NSCLC is 9.5 months.
Adenovirus
Adenovirus is a non-enveloped, double-stranded DNA virus with a large linear genome (~ 35 Kb) encapsulated by an icosahedral capsid [1•]. Due to the large genome, long DNA sequences can be incorporated, thus permitting multiple engineered modifications. Upon cell entry, an adenovirus moves to the nucleus where it expresses adenoviral early genes (encoding E1A and E1B), which are necessary for viral propagation. E1A and E1B target the tumor suppressors p53 and pRb to promote cell cycle entry. In normal host cells, the targeting of host p53 and pRb by the adenoviral E1A and E1B proteins results in apoptosis and clearance of the virus [40, 41]. As adenoviruses lacking the E1b protein may specifically replicate in tumor cells that do not express active p53, ONYX-015 and H101 have a deletion in the portion of E1B that inactivates p53.
Wild-type p53 gene can be introduced into human tumor cells with a mutant p53 genotype using adenovirus [42]. To study the benefit from adenoviral p53 (Adp53) gene therapy in first-line setting [43], 25 patients with unresectable NSCLC were enrolled in an open-label, multicenter phase II study with three cycles of carboplatin and paclitaxel or cisplatin and vinorelbine in combination with intratumoral injection of 7.5 × 1012 particles of SCH 58,500 (rAd/p53, day 1 of each cycle). No difference was found between the response rate of lesions treated with p53 gene therapy with chemotherapy (52%) and lesions treated with chemotherapy alone (48%). There was also no survival difference between the two chemotherapy regimens (1-year survival, 44%). Transgene expression was confirmed in 68% of the tumor samples. Treatment-related adverse events were mild to moderate. In a phase I trial [44], adenovirus vector containing wild-type p53 complementary DNA (Ad-p53) was administered to 28 patients with pretreated refractory NSCLC. Patients received up to six, monthly intratumoral injections of Ad-p53 by use of computed tomography-guided percutaneous fine-needle injection (23 patients) or bronchoscopy (five patients). Polymerase chain reaction (PCR) analysis showed the presence of adenovirus vector DNA in 18 (86%) of 21 patients with evaluable post-treatment biopsy specimens; vector-specific p53 messenger RNA (mRNA) was detected by real time PCR (RT-PCR) analysis in 12 (46%) of 26 patients. Apoptosis was confirmed in post-treatment biopsy specimens from 11 patients. Treatment was well tolerated and two (8%) patients had partial response and an additional 16 (64%) patients had stable disease lasting 2 to 14 months. This trial thus confirmed transgene expression of wild-type p53, and showed modest antitumor activity in advanced NSCLC. Despite transgene p53 expression in tumor cells, intratumoral adenoviral p53 gene therapy appears to provide no additional benefit in patients receiving first-line chemotherapy for advanced NSCLC.
A phase 2 study investigated the addition of stereotactic body radiation therapy (SBRT) and intratumoral injection of the oncolytic virus ADV/HSV-tk (adenovirus-mediated expression of herpes simplex virus thymidine kinase) to PD-1 antibody for the treatment of stage IV NSCLC [45]. Patients (both IO-naïve and IO-resistant) received an intratumoral injection of ADV/HSV-tk (5 × 1011 vp) followed by SBRT (30 Gy in 5 fractions) to the same tumor. Pembrolizumab or Nivolumab (anti-PD-1 agents) was given for up to 2 years. The ORR and clinical benefit rate (CBR) were 28.5% and 61.9% in the immunotherapy-naive group, and 14.2% and 64.2% in the group that previously received IO, respectively. Gene-mediated cytotoxic immunotherapy (GMCI) is an immunotherapuetic approach in which adenovirus-based vector expressing the thymidine kinase gene (aglatimagene besadenovec, AdV-tk) is locally administered followed by anti-herpetic prodrug valacyclovir [46]. A phase I dose escalation trial of GMCI followed by chemotherapy has been completed in patients with malignant pleural effusion. AdV-tk was administered intrapleurally in three cohorts at a dose of 1 × 1012 to 1013 vector particles. Nineteen patients were enrolled: 14 with malignant mesothelioma, 4 NSCLC, and 1 breast cancer. There were no DLTs. Three patients experienced transient cytokine release syndrome. Three of four patients with NSCLC had durable stabilization of disease with one patient still in follow up 29 months after therapy. A phase II study of GMCI in combination with standard of care immune checkpoint inhibitor for patients with IO-resistant NSCLC is currently enrolling (NCT04495153).
Seneca Valley Virus
Seneca valley virus is an oncolytic, non-enveloped RNA virus (picornavirus) with tropism for small cell lung cancer (SCLC). It can be delivered intravenously because of its natural resistance to hemagglutination, a process that usually results in premature viral clearance and reduced delivery to the tumor site following intravenous delivery [47••]. During replication, it does not have a DNA phase and cannot be incorporated into the host genome. In a phase 1 trial, 30 patients with advanced solid tumors were treated with Seneca vally virus (SVV-001), including six with SCLC [48]. SVV-001 was well tolerated and there were no DLTs. Viral clearance was confirmed in all patients and correlated with development of antiviral antibodies. Evidence of in vivo intratumoral viral replication was observed in patients with SCLC, with peak viral titers estimated to be > 103-fold higher than the administered dose. One patient with previously chemo-resistant SCLC remained progression-free for 10 months. A phase II double-blind, placebo-controlled trial evaluated Seneca valley virus (NTX-010) in patients with extensive-stage (ES) SCLC after completion of first-line chemotherapy [47••]. From 2010 to 2013, 50 patients were randomized 1:1 to a single dose of NTX-010 or placebo within 12 weeks of chemotherapy. The primary end point was progression free survival (PFS). At the specified interim analysis, the median PFS was 1.7 months for the NTX-010 group versus 1.7 months for the placebo group (HR 1.03, p = 0.92), and the trial was terminated owing to futility. Patients in whom NTX-010 persisted in the blood one to two weeks after treatment had a shorter PFS. Rate of grade 3 or higher adverse events, regardless of attribution, was 34.6% with the common adverse events being flu like symptoms, diarrhea, and fatigue.
Reovirus
Reovirus are double-stranded, non-enveloped RNA viruses that show natural selectivity for cancer cells with an over-active RAS signaling pathway. In normal healthy cells, reovirus is able to enter the cell and begin producing viral RNAs in the cytoplasm, which activates the protein kinase to double-stranded RNA (PKR) pathway. Activated PKR, in turn, inhibits protein translation, preventing the production of viral particles and stopping the spread of the virus [49]. However, RAS-transformed cancer cells do not initiate the PKR pathway, making these tumor cells permissive to infection and cell lysis [50]. The drawback is it cannot kill tumor cells that do not have an activated RAS pathway. There is also preclinical data to support that reovirus is synergistic in combination with chemotherapy for patients with NSCLC [51].
Pelareorep, a Dearing strain of reovirus serotype 3, was tested in a randomized phase II trial through the Canadian Cancer Trial group that enrolled patients with advanced or metastatic NSCLC after progression on first line chemotherapy [52]. Patients were randomized 1:1 to chemotherapy (pemetrexed or docetaxel based on histology) with or without pelareorep (4.5 × 1010 TCID50, days 1 to 3 every 21 days). The primary outcome was PFS. Between 2012 and 2015, 166 patients were enrolled. Pelareorep did not improve the PFS in the pelareorep arm versus single agent chemotherapy (median PFS 3.0 months vs. 2.8 months, HR 0.90, p = 0.53). Neither the presence of a KRAS nor EGFR mutation was associated with improved PFS. The combination was tolerable, although associated with increased rates of neutropenic fever. Another study evaluated Reolysin (type 3 dearing reovirus) [53••] combined with paclitaxel and carboplatin in patients with metastatic or recurrent KRAS-mutated or EGFR-mutated/amplified NSCLC in first line setting. Thirty-seven patients were treated and Reolysin was administered on days 1 to 5 of each 21-day cycle. ORR was 31%, median PFS was 4 months, and median OS was 12 months. The combination therapy was well tolerated but the lack of a comparator arm limited interpretation of the efficacy data.
Vaccinia Virus
Vaccinia virus, a member of the poxvirus family, has a large double stranded DNA (dsDNA) genome (~ 190 kb) [1•]. The large genome enables insertion of a large amount of foreign DNA without significantly reducing the replication ability of the virus [54]. Replication occurs entirely in the cytoplasm eliminating concerns about insertional mutagenesis. Vaccinia virus is also highly tropic for tumor cells [55]. TG4010 (MVA-MUC1-IL-2) is a vector-based vaccine targeting the tumor associated MUC1 antigen expressed by tumors [56]. It consists of a viral suspension of Ankara virus, an attenuated, genetically modified vaccinia virus, to express MUC-1 and IL-2. In a phase I dose escalation trial [57], 108 PFU per injection was determined to be maximal feasible dose. Three of the patients in this trial had metastatic NSCLC with one experiencing reduction in multiple tumor sites with a two months delay after the last vaccination.
A phase IIB randomized, double-blind, placebo-controlled, multicenter phase trial explored two schedules of the combination of TG4010 with first-line chemotherapy in 65 patients with stage IIIB/IV NSCLC [58]. In Arm 1, TG4010 was combined upfront with cisplatin and vinorelbine. In Arm 2, patients were treated with TG4010 monotherapy until disease progression, followed by TG4010 plus the same chemotherapy as in Arm 1. In Arm 1, partial response (PR) was observed in 13 out of 37 evaluable patients (35.1%). However, Arm 2 did not meet the criteria for moving forward to second stage as there were no responses with TG4010 alone. The median OS was 12.7 months in Arm 1 and 14.9 months in Arm 2. TG4010 was well tolerated, common adverse events were injection site reactions, flu-like symptoms, and fatigue. A MUC1-specific cellular immune response was observed in lymphocyte samples from responding patients. Another phase IIb study assessed TG4010 in combination with first-line chemotherapy in 148 patients with advanced NSCLC expressing MUC1 by IHC [59]. Patients were randomized to TG4010 in combination with cisplatin and gemcitabine or to chemotherapy alone. The primary outcome, 6-month PFS was 43.2% in the combination group compared to 35.1% in the chemotherapy alone group (p = 0.31). The time to progression and median OS were also not significantly different between the two groups.
In the phase IIb part of the phase IIb/III randomized TIME trial (NCT01383148) [60••], 222 patients with advanced NSCLC were randomized to TG4010 and chemotherapy versus placebo and chemotherapy in the first-line setting. The primary endpoint was PFS, and the correlative endpoint was to validate the baseline value of CD16, CD56, and CD69 triple-positive activated lymphocytes (TrPAL) as a predictive biomarker. Median PFS was 5.9 months in the TG4010 group and 5.1 months in the placebo group (HR 0.74; one-sided p = 0.019). In patients with TrPAL values of less than or equal to the upper limit of normal (ULN), the HR for PFS was 0.75 (0.54–1.03), and thus the primary endpoint was met. No serious adverse events were reported as related to the combination treatment. Subsequently, a phase II trial evaluating TG4010 in combination with chemotherapy and nivolumab for first-line treatment of advanced NSCLC and PD-L1 < 50% was initiated (NCT03353675). However, in 2019, it was reported that the trial did not meeting the primary endpoint (ORR) and further development of TG4010 was discontinued.
Challenges to Developing Oncolytic Viruses in Lung Cancer
Viral delivery remains a major issue for development of OVs for thoracic cancers as many OVs can only be administered directly into the tumor. Vaccinia and reovirus are good viruses for systemic delivery; however, the doses required to achieve systemic tumor replication are high and close to the limits of production capacity [61]. One approach to overcome this challenge is to use carrier cells for the targeted, systemic delivery of OVs. By combining OVs with immune cell-based carrier systems, the opportunity to improve the bioavailability of OVs can be combined with adoptive cell transfer for increased effectiveness of the therapy [62]. Various agents such as nanoparticles, liposomes, polyethylene glycol, and polymeric particles have been used to deliver OVs from the systemic circulation to the local tumor bed [63, 64]. Magnetic drug targeted systems have also become a promising carrier system to effectively deliver viruses to tumor cells [65].
Preexisting neutralizing antiviral antibodies are another challenge for systemic delivery of free virus to reach the tumor bed [66]. Potential strategies to address this are using alternative virus serotypes, PEGylation of viral coat, polymer coating to prevent antibody binding, and neutralization [1•]. Also, limited efficacy with OVs observed so far with lung cancer may be due to variable viral doses reaching the tumor as OVs are live viruses and proliferate only after systemic administration [67]. Efforts should focus on maximizing the effective viral load in tumor lesions through improved tumor selectivity, thereby improving efficacy, which is also based on improved tumor selectivity [68].
New Combinations in Development with Oncolytic Viruses
A number of novel approaches are being explored to improve the efficacy of OVs. The combination of OVs with tumor antigen-targeting vaccines has the potential to activate and amplify tumor-reactive cytotoxic T-cells (CTLs) through OV-mediated immunoadjuvant effects [69]. Mixture of a DC1 vaccination and oncolytic vaccinia virus expressing CCL5 (vvCCL5; receptors that are expressed on CTLs induced by DC1) induced chemotaxis of lymphocyte populations both in vitro and in vivo, and showed enhanced antitumor effect in tumor-bearing mice vaccinated with DC1 and treated with vvCCL5 [70]. The concept of pre-infection using adenovirus and oncolytic vaccinia virus as a boost prior to vaccine delivery to improve vaccine immunogenicity has been confirmed in other preclinical studies as well [71, 72]. OVs can also be synergistic with Chimeric antigen receptor T (CAR-T) cell therapy. In a mouse neuroblastoma model, oncolytic adenovirus with the chemokine RANTES and the cytokine IL-15 (Ad5Δ24) enhanced migration and proliferation of CAR-T cells [73]. The combination therapy of Ad5Δ24 and CAR-T cells also increased the OS of tumor-bearing mice [73]. Bispecific T cell engagers (BiTEs) are a subclass of bispecific antibodies that have specific antibodies for CD3 on one arm and another antibody for a tumor antigen on the second arm [74]. Wing et al. generated an oncolytic adenovirus armed with an EGFR-targeting BiTE (OAd-BiTE) and demonstrated that OAd-BiTE with EGFR-targeting CAR-T therapy improved anti-tumor efficacy and prolonged survival in various mouse cancer models [75].
Conclusion
In conclusion, OVs have to date demonstrated a tolerable safety profile as well as ability to modify the tumor microenvironment and kill tumor cells. However, for lung cancer, development of OVs has been limited due to difficulty in achieving adequate viral load at all tumor sites and limited benefit from OV monotherapy. Combination therapy of OVs with chemotherapy showed moderate benefit in early phase trials but were not validated in larger, randomized trials. Developing novel combinations with OVs, especially with checkpoint inhibitors and other immunotherapeutics, may be a strategy to address the limited success seen so far. However, integrating appropriate biomarkers studies as well as meaningful endpoints in future clinical trials will be key to the future development of OVs for lung cancer. Using novel viral delivery systems may be another strategy to improve OV efficacy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2016;15(9):660. https://doi.org/10.1038/nrd.2016.178. This review summarizes the appoach to development of oncolytic viruses for cancer therapy.
Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther Oncolytics. 2019;15:234–47. https://doi.org/10.1016/j.omto.2019.10.007.
Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, Vile RG. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20(10):1119–32. https://doi.org/10.1089/hum.2009.135.
Di Paolo NC, Miao EA, Iwakura Y, Murali-Krishna K, Aderem A, Flavell RA, Papayannopoulou T, Shayakhmetov DM. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31(1):110–21. https://doi.org/10.1016/j.immuni.2009.04.015.
Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir(R) story. Eur J Pharmacol. 2018;837:117–26. https://doi.org/10.1016/j.ejphar.2018.08.042.
Liang M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr Cancer Drug Targets. 2018;18(2):171–6. https://doi.org/10.2174/1568009618666171129221503.
Zeng J, Li X, Sander M, Zhang H, Yan G, Lin Y. Oncolytic viro-immunotherapy: an emerging option in the treatment of gliomas. Front Immunol. 2021;12:721830. https://doi.org/10.3389/fimmu.2021.721830.
• Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH Jr, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8. https://doi.org/10.1200/JCO.2014.58.3377. This trial presented data to support FDA approval of talimogene laherparevec for the treatment of advanced melanoma.
• Ekeke CN, Russell KL, Joubert K, Bartlett DL, Luketich JD, Soloff AC, Guo ZS, Lotze MT, Dhupar R. Fighting fire with fire: oncolytic virotherapy for thoracic malignancies. Ann Surg Oncol. 2021;28(5):2715–27. https://doi.org/10.1245/s10434-020-09477-4. This articles describes the approach to development of oncolytic viruses for the treatment of lung cancer.
Bai Y, Hui P, Du X, Su X. Updates to the antitumor mechanism of oncolytic virus. Thorac Cancer. 2019;10(5):1031–5. https://doi.org/10.1111/1759-7714.13043.
Matsuda T, Karube H, Aruga A. A comparative safety profile assessment of oncolytic virus therapy based on clinical trials. Ther Innov Regul Sci. 2018;52(4):430–7. https://doi.org/10.1177/2168479017738979.
Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-001486.
Wang Y, Zhou X, Wu Z, Hu H, Jin J, Hu Y, Dong Y, Zou J, Mao Z, Shi X, Huo Y, Lyu J, Fang Z, Zhang W, Zhu Y, Li B, Liu B. Preclinical safety evaluation of oncolytic herpes simplex virus type 2. Hum Gene Ther. 2019;30(5):651–60. https://doi.org/10.1089/hum.2018.170.
Kimball KJ, Preuss MA, Barnes MN, Wang M, Siegal GP, Wan W, Kuo H, Saddekni S, Stockard CR, Grizzle WE, Harris RD, Aurigemma R, Curiel DT, Alvarez RD. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin Cancer Res. 2010;16(21):5277–87. https://doi.org/10.1158/1078-0432.CCR-10-0791.
Wei N, Fan JK, Gu JF, He LF, Tang WH, Cao X, Liu XY. A double-regulated oncolytic adenovirus with improved safety for adenocarcinoma therapy. Biochem Biophys Res Commun. 2009;388(2):234–9. https://doi.org/10.1016/j.bbrc.2009.07.142.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62. https://doi.org/10.1038/nrd4663.
Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107(10):1373–9. https://doi.org/10.1111/cas.13027.
Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74. https://doi.org/10.3389/fonc.2014.00074.
Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4(6):e1008824. https://doi.org/10.1080/2162402X.2015.1008824.
Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, Brunelliere J, Bramson JL, Lichty BD, Wan Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18(8):1430–9. https://doi.org/10.1038/mt.2010.98.
Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tahtinen S, Oksanen M, Heiskanen R, Pesonen S, Joensuu T, Alanko T, Partanen K, Laasonen L, Kairemo K, Pesonen S, Kangasniemi L, Hemminki A. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19(10):2734–44. https://doi.org/10.1158/1078-0432.CCR-12-2546.
Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, Kubicka S, Kuhnel F. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23(10):1630–40. https://doi.org/10.1038/mt.2015.115.
Reale A, Vitiello A, Conciatori V, Parolin C, Calistri A, Palu G. Perspectives on immunotherapy via oncolytic viruses. Infect Agent Cancer. 2019;14:5. https://doi.org/10.1186/s13027-018-0218-1.
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–6. https://doi.org/10.1126/science.1851332.
Pikor LA, Bell JC, Diallo JS. Oncolytic viruses: exploiting cancer’s deal with the devil. Trends Cancer. 2015;1(4):266–77. https://doi.org/10.1016/j.trecan.2015.10.004.
Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. 2020;11:683. https://doi.org/10.3389/fimmu.2020.00683.
Wang LC, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK, Sun J, Fridlender ZG, Isaacs SN, Thorne SH, Albelda SM. Treating tumors with a vaccinia virus expressing IFNbeta illustrates the complex relationships between oncolytic ability and immunogenicity. Mol Ther. 2012;20(4):736–48. https://doi.org/10.1038/mt.2011.228.
Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, Kottke T, Federspiel M, Barber G, Albelda SM, Vile RG. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009;69(19):7713–20. https://doi.org/10.1158/0008-5472.CAN-09-1013.
Sterman DH, Recio A, Carroll RG, Gillespie CT, Haas A, Vachani A, Kapoor V, Sun J, Hodinka R, Brown JL, Corbley MJ, Parr M, Ho M, Pastan I, Machuzak M, Benedict W, Zhang XQ, Lord EM, Litzky LA, Heitjan DF, June CH, Kaiser LR, Vonderheide RH, Albelda SM, Kanther M. A phase I clinical trial of single-dose intrapleural IFN-beta gene transfer for malignant pleural mesothelioma and metastatic pleural effusions: high rate of antitumor immune responses. Clin Cancer Res. 2007;13(15 Pt 1):4456–66. https://doi.org/10.1158/1078-0432.CCR-07-0403.
Corke L, Sacher A. New strategies and combinations to improve outcomes in immunotherapy in metastatic non-small-cell lung cancer. Curr Oncol. 2021;29(1):38–55. https://doi.org/10.3390/curroncol29010004.
Zhang H, Xie W, Zhang Y, Dong X, Liu C, Yi J, Zhang S, Wen C, Zheng L, Wang H. Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther. 2021. https://doi.org/10.1038/s41417-021-00389-3.
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, Kirkwood JM, Gajewski TF, Chen L, Gorski KS, Anderson AA, Diede SJ, Lassman ME, Gansert J, Hodi FS, Long GV. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109-19 e10. https://doi.org/10.1016/j.cell.2017.08.027.
Shafren DR, Dorahy DJ, Ingham RA, Burns GF, Barry RD. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol. 1997;71(6):4736–43. https://doi.org/10.1128/JVI.71.6.4736-4743.1997.
Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K, Nakanishi Y, Shimizu H, Tani K. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72(10):2609–21. https://doi.org/10.1158/0008-5472.CAN-11-3185.
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FA, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118(1):209–14. https://doi.org/10.1002/ijc.21290.
Guo WF, Lin RX, Huang J, Zhou Z, Yang J, Guo GZ, Wang SQ. Identification of differentially expressed genes contributing to radioresistance in lung cancer cells using microarray analysis. Radiat Res. 2005;164(1):27–35. https://doi.org/10.1667/rr3401.
Deng H, Liu H, de Silva T, Xue Y, Mohamud Y, Ng CS, Qu J, Zhang J, Jia WWG, Lockwood WW, Luo H. Coxsackievirus type B3 is a potent oncolytic virus against KRAS-mutant lung adenocarcinoma. Mol Ther Oncolytics. 2019;14:266–78. https://doi.org/10.1016/j.omto.2019.07.003.
Pandha H, Harrington K, Ralph C, Melcher A, Gupta S, Akerley W, Sandborn RE, Rudin C, Rosenberg J, Kaufman D, Schmidt E, Grose M, Shafren DR. Phase 1b KEYNOTE 200 (STORM study): A study of an intravenously delivered oncolytic virus, Coxsackievirus A21 in combination with pembrolizumab in advanced cancer patients [abstract]. Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1–5; Washington, DC Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract nr CT115
Rudin CM, Pandha HS, Gupta S, Zibelman MR, Akerley W, Day D, Hill AG, Sanborn RE, O’Day SJ, Clay TD, Wright GM, Jennens R, Gerber DE, Rosenberg JE, Ralph C, Campbell DC, Curti BD, Schmidt EV, Grose M, Shafen D. Phase Ib KEYNOTE-200: A study of an intravenously delivered oncolytic virus, coxsackievirus A21 in combination with pembrolizumab in advanced NSCLC and bladder cancer patients. Ann Oncol. 2018;29(8):VIII732.
Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–6. https://doi.org/10.1126/science.274.5286.373.
Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci U S A. 1992;89(16):7742–6. https://doi.org/10.1073/pnas.89.16.7742.
Nemunaitis J, Swisher SG, Timmons T, Connors D, Mack M, Doerksen L, Weill D, Wait J, Lawrence DD, Kemp BL, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie JM, Lee JJ, Lee JS, Nguyen DM, Nesbitt JC, Perez-Soler R, Pisters KM, Putnam JB, Richli WR, Shin DM, Walsh GL, Merritt J, Roth J. Adenovirus-mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. J Clin Oncol. 2000;18(3):609–22. https://doi.org/10.1200/JCO.2000.18.3.609.
Schuler M, Herrmann R, De Greve JL, Stewart AK, Gatzemeier U, Stewart DJ, Laufman L, Gralla R, Kuball J, Buhl R, Heussel CP, Kommoss F, Perruchoud AP, Shepherd FA, Fritz MA, Horowitz JA, Huber C, Rochlitz C. Adenovirus-mediated wild-type p53 gene transfer in patients receiving chemotherapy for advanced non-small-cell lung cancer: results of a multicenter phase II study. J Clin Oncol. 2001;19(6):1750–8. https://doi.org/10.1200/JCO.2001.19.6.1750.
Swisher SG, Roth JA, Nemunaitis J, Lawrence DD, Kemp BL, Carrasco CH, Connors DG, El-Naggar AK, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie JM, Lee JJ, Lee JS, Mack M, Merritt JA, Nguyen DM, Nesbitt JC, Perez-Soler R, Pisters KM, Putnam JB Jr, Richli WR, Savin M, Schrump DS, Shin DM, Shulkin A, Walsh GL, Wait J, Weill D, Waugh MK. Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst. 1999;91(9):763–71. https://doi.org/10.1093/jnci/91.9.763.
Guerrero C, Ensor JE, Sun K, Farach AM, Nair S, Zhang J, Singh M, Darcourt JG, Ramshesh PV, Butler EB, Teh BS, Sultenfuss M, Gupta N, Heslop HE, Mejia JA, Chang JC, Bernicker E. Stereotactic body radiation therapy and in situ oncolytic virus therapy followed by immunotherapy in metastatic non-small cell lung cancer. J Clin Oncol. 2021;39(15_suppl):9115.
Aggarwal C, Haas AR, Metzger S, Aguilar LK, Aguilar-Cordova E, Manzanera AG, Gomez-Hernandez G, Katz SI, Alley EW, Evans TL, Bauml JM, Cohen RB, Langer CJ, Albelda SM, Sterman DH. Phase I study of intrapleural gene-mediated cytotoxic immunotherapy in patients with malignant pleural effusion. Mol Ther. 2018;26(5):1198–205. https://doi.org/10.1016/j.ymthe.2018.02.015.
•• Schenk EL, Mandrekar SJ, Dy GK, Aubry MC, Tan AD, Dakhil SR, Sachs BA, Nieva JJ, Bertino E, Lee Hann C, Schild SE, Wadsworth TW, Adjei AA, Molina JR. A randomized double-blind phase II study of the Seneca Valley Virus (NTX-010) versus placebo for patients with extensive-stage SCLC (ES SCLC) who were stable or responding after at least four cycles of platinum-based chemotherapy: North Central Cancer Treatment Group (Alliance) N0923 Study. J Thorac Oncol. 2020;15(1):110–9. https://doi.org/10.1016/j.jtho.2019.09.083. This is one of the largest trials for the treatment of small cell lung cancer using oncolytic viruses.
Rudin CM, Poirier JT, Senzer NN, Stephenson J Jr, Loesch D, Burroughs KD, Reddy PS, Hann CL, Hallenbeck PL. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res. 2011;17(4):888–95. https://doi.org/10.1158/1078-0432.CCR-10-1706.
Bischoff JR, Samuel CE. Mechanism of interferon action. Activation of the human P1/eIF-2 alpha protein kinase by individual reovirus s-class mRNAs s1 mRNA is a potent activator relative to s4 mRNA. Virology. 1989;172(1):106–15. https://doi.org/10.1016/0042-6822(89)90112-8.
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351–62. https://doi.org/10.1093/emboj/17.12.3351.
Sei S, Mussio JK, Yang QE, Nagashima K, Parchment RE, Coffey MC, Shoemaker RH, Tomaszewski JE. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer. 2009;8:47. https://doi.org/10.1186/1476-4598-8-47.
Bradbury PA, Morris DG, Nicholas G, Tu D, Tehfe M, Goffin JR, Shepherd FA, Gregg RW, Rothenstein J, Lee C, Kuruvilla S, Keith BD, Torri V, Blais N, Hao D, Korpanty GJ, Goss G, Melosky BL, Mates M, Leighl N, Ayoub JP, Sederias J, Feilotter H, Seymour L, Laurie SA. Canadian Cancer Trials Group (CCTG) IND211: A randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer. 2018;120:142–8. https://doi.org/10.1016/j.lungcan.2018.03.005.
•• Villalona-Calero MA, Lam E, Otterson GA, Zhao W, Timmons M, Subramaniam D, Hade EM, Gill GM, Coffey M, Selvaggi G, Bertino E, Chao B, Knopp MV. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer. 2016;122(6):875–83. https://doi.org/10.1002/cncr.29856. This trial is an early-phase trial reporting results from combination oncolytic reovirus and chemotherapy for patients with non-small cell lung cancer.
Truong CS, Yoo SY. Oncolytic vaccinia virus in lung cancer vaccines. Vaccines (Basel). 2022;10(2). https://doi.org/10.3390/vaccines10020240.
Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, Diallo JS, Falls T, Burns J, Garcia V, Kanji F, Evgin L, Hu K, Paradis F, Knowles S, Hwang TH, Vanderhyden BC, Auer R, Kirn DH, Bell JC. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749–58. https://doi.org/10.1038/mt.2011.276.
Cohen S, Kaufman HL. TG-4010 Transgene. Curr Opin Investig Drugs. 2004;5(12):1319–28.
Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, Tartour E, Zhao Y, Bizouarne N, Baudin M, Acres B. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med. 2003;5(8):690–9. https://doi.org/10.1002/jgm.397.
Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Levy E, Krzakowski M, Hess D, Tartour E, Chenard MP, Limacher JM, Bizouarne N, Acres B, Halluard C, Velu T. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol. 2008;3(7):735–44. https://doi.org/10.1097/JTO.0b013e31817c6b4f.
Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, Koralewski P, Breton JL, Stoelben E, Braun D, Debieuvre D, Lena H, Buyse M, Chenard MP, Acres B, Lacoste G, Bastien B, Tavernaro A, Bizouarne N, Bonnefoy JY, Limacher JM. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 2011;12(12):1125–33. https://doi.org/10.1016/S1470-2045(11)70259-5.
•• Quoix E, Lena H, Losonczy G, Forget F, Chouaid C, Papai Z, Gervais R, Ottensmeier C, Szczesna A, Kazarnowicz A, Beck JT, Westeel V, Felip E, Debieuvre D, Madroszyk A, Adam J, Lacoste G, Tavernaro A, Bastien B, Halluard C, Palanche T, Limacher JM. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016;17(2):212–23. https://doi.org/10.1016/S1470-2045(15)00483-0. This trial is one of the largest phase III vaccine trial completed for the treatment of lung cancer.
Dash AS, Patel MR. Viroimmunotherapy of thoracic cancers. Biomedicines. 2017;5(1). https://doi.org/10.3390/biomedicines5010002
Reale A, Calistri A, Altomonte J. Giving oncolytic viruses a free ride: carrier cells for oncolytic virotherapy. pharmaceutics. 2021;13(12). https://doi.org/10.3390/pharmaceutics13122192.
Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009;20(9):975–88. https://doi.org/10.1089/hum.2009.028.
Green NK, Herbert CW, Hale SJ, Hale AB, Mautner V, Harkins R, Hermiston T, Ulbrich K, Fisher KD, Seymour LW. Extended plasma circulation time and decreased toxicity of polymer-coated adenovirus. Gene Ther. 2004;11(16):1256–63. https://doi.org/10.1038/sj.gt.3302295.
Hill C, Carlisle R. Achieving systemic delivery of oncolytic viruses. Expert Opin Drug Deliv. 2019;16(6):607–20. https://doi.org/10.1080/17425247.2019.1617269.
Liu XQ, Xin HY, Lyu YN, Ma ZW, Peng XC, Xiang Y, Wang YY, Wu ZJ, Cheng JT, Ji JF, Zhong JX, Ren BX, Wang XW, Xin HW. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018;25(1):1950–62. https://doi.org/10.1080/10717544.2018.1534895.
Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18(8):498–513. https://doi.org/10.1038/s41577-018-0014-6.
Li L, Liu S, Han D, Tang B, Ma J. Delivery and biosafety of oncolytic virotherapy. Front Oncol. 2020;10:475. https://doi.org/10.3389/fonc.2020.00475.
Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018;33(4):599–605. https://doi.org/10.1016/j.ccell.2018.03.011.
Li J, O’Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, Thorne SH, Bartlett DL. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19(4):650–7. https://doi.org/10.1038/mt.2010.312.
Lu S, Zhang Z, Du P, Chard LS, Yan W, El Khouri M, Wang Z, Zhang Z, Chu Y, Gao D, Zhang Q, Zhang L, Nagano A, Wang J, Chelala C, Liu J, Chen J, Liu P, Dong Y, Wang S, Li X, Dong J, Lemoine NR, Pei D, Wang Y. A virus-infected, reprogrammed somatic cell-derived tumor cell (VIReST) vaccination regime can prevent initiation and progression of pancreatic cancer. Clin Cancer Res. 2020;26(2):465–76. https://doi.org/10.1158/1078-0432.CCR-19-1395.
Tysome JR, Li X, Wang S, Wang P, Gao D, Du P, Chen D, Gangeswaran R, Chard LS, Yuan M, Alusi G, Lemoine NR, Wang Y. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin Cancer Res. 2012;18(24):6679–89. https://doi.org/10.1158/1078-0432.CCR-12-0979.
Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74(18):5195–205. https://doi.org/10.1158/0008-5472.CAN-14-0697.
Suryadevara CM, Gedeon PC, Sanchez-Perez L, Verla T, Alvarez-Breckenridge C, Choi BD, Fecci PE, Sampson JH. Are BiTEs the “missing link” in cancer therapy? Oncoimmunology. 2015;4(6):e1008339. https://doi.org/10.1080/2162402X.2015.1008339.
Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T, Young RM, Alemany R, June CH, Guedan S. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6(5):605–16. https://doi.org/10.1158/2326-6066.CIR-17-0314.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jyoti Malhotra has served on the advisory board for Astra-Zeneca, Blueprint Medicines, Mirati Therapeutics, Sanofi, Oncocyte, and Beigene and received research funding from Bristol-Myers Squibb, Celldex, Biohaven, Daiichi Sankyo, and Beyond Spring Pharmaceuticals. Edward Kim has received personal fees from AstraZeneca and Genentech.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lung Cancer
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Malhotra, J., Kim, E.S. Oncolytic Viruses and Cancer Immunotherapy. Curr Oncol Rep 25, 19–28 (2023). https://doi.org/10.1007/s11912-022-01341-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-022-01341-w