
Abstract
Purpose of Review
Apoptosis is a major mechanism of cancer cell death. Thus, evasion of apoptosis results in therapy resistance. Here, we review apoptosis modulators in cancer and their recent developments, including MDM2 inhibitors and kinase inhibitors that can induce effective apoptosis.
Recent Findings
Both extrinsic pathways (external stimuli through cell surface death receptor) and intrinsic pathways (mitochondrial-mediated regulation upon genotoxic stress) regulate the complex process of apoptosis through orchestration of various proteins such as members of the BCL-2 family. Dysregulation within these complex steps can result in evasion of apoptosis. However, via the combined evolution of medicinal chemistry and molecular biology, omics assays have led to innovative inducers of apoptosis and inhibitors of anti-apoptotic regulators. Many of these agents are now being tested in cancer patients in early-phase trials.
Summary
We believe that despite a sluggish speed of development, apoptosis targeting holds promise as a relevant strategy in cancer therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
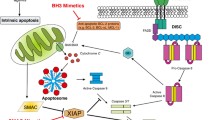
Apoptosis is a programmed cell death process utilized by normal and cancer cells, along with necroptosis, ferroptosis, autophagy, pyroptosis, and others [1, 1]. Apoptosis is a specific form of programmed cell death that involves the blebbing of the cell membrane, which exposes the phosphatidylserine, as well as activates cysteine-aspartic protease (caspase) family proteins [2]. Like many sophisticated regulatory pathways, apoptosis can be a double-edged sword in cancer progression. For example, while apoptosis due to anti-cancer therapy can kill cancer cells, sublethal activation of caspase 3 may result in oncogenic progression, since suboptimal induction of apoptosis can trigger compensatory survival mechanisms such as autophagy [3]. Hence, it is challenging to find the delicate balance between effective apoptosis and inefficient apoptotic pathway activation, leading to further progression of cancer [4]. The current mainstay of cancer treatment is still cytotoxic chemotherapy. Yet, many patients treated with chemotherapy develop residual tumor or recurrent metastatic disease [5, 6, 7]. As apoptosis is critical for maintaining homeostasis in normal cells, defects in apoptosis help cancer cells escape (Fig. 8), causing resistance to standard therapy. The apoptosis pathway also cross-talks with many growth factors and other growth-mediating pathways; therefore, dysregulation of apoptosis can promote tumor growth and progression [9]. A better strategy to induce effective cell death at the initial stage of treatment is essential.

How evasion of apoptosis can help cancer cells to survive through chemotherapy, radiation therapy, and immune checkpoint inhibitor therapies. Apoptosis = targeted inhibitors can be used either in combination with standard therapies or can be used to treat residual cancer cells when standard treatments are not effective
There are two main apoptotic regulation pathways: extrinsic and intrinsic. The extrinsic pathway is triggered by the binding of tumor necrosis factor (TNF), TNF-related apoptosis-inducing ligand (TRAIL), or FAS ligand (APO-1) to the corresponding receptor (TNFR, TRAILR, or FAS, respectively) [10, 11]. Once triggered, activated caspase 8 truncates the BID protein to truncated BID, subsequently activating the pro-apoptotic regulators BAX and BAK. This activation induces mitochondrial outer membrane permeabilization (MOMP) and stimulates the beginning of intrinsic pathway and caspase activation [12, 13]. The intrinsic pathway is also triggered by genotoxic stress to the cells, via unfolded protein response, reactive oxygen species, radiation, and chemical-induced chromosomal abnormalities [14, 15]. MOMP leads to a release and activation of second mitochondria-derived activator of caspase (Smac), also called direct inhibitor of apoptosis protein [IAP]-binding protein with low pI (DIABLO), from the intermembrane space of mitochondria into the cytosol [16] and release of cytochrome C. Released cytochrome C then assembles the apoptosome along with apoptotic peptidase activating factor 1 (APAF1), dATP, and pro-caspase 9 in the cytosol. Smac/DIABLO inactivates anti-apoptotic regulator IAP proteins (XIAP, cIAP1, and cIAP2), resulting in apoptosis. BH3 domain-containing molecules are also essential regulators of the intrinsic pathway; these molecules include BID, BAX, BAK, and BCL-2 family members [17, 18]. Recently, leucine zipper kinases also have been increasingly recognized as key regulators of apoptosis that can be therapeutically targeted.
Type I apoptotic cells induce apoptosis extrinsically independent of mitochondria, while type II apoptotic cells rely on intrinsic, mitochondrial pathways for efficient cell death [19, 20]. Epithelial cells are often type II, while other cells may be type I. Therefore, cancer cells that are of epithelial cell origin most likely utilize both extrinsic and intrinsic apoptosis pathways [21].
The complex regulation of apoptosis creates considerable barriers for drug developers to harness apoptosis induction in effective anti-cancer therapeutics. Nonetheless, the rapidly evolving field of medical chemistry along with omics technologies and translational abilities has produced new apoptosis-targeted agents that are undergoing clinical development. Only a handful of these agents have been approved by the US Food and Drug Administration (FDA) for clinical use within the last decade, indicating that considerable effort is needed to develop apoptosis-targeted therapeutics as a viable strategy.
Extrinsic Pathway and Targeted Agents
The extrinsic apoptosis pathway is initiated and targeted at the cell surface receptor level upstream of intrinsic apoptosis regulation, although the pathway used depends on the cell type. A main trigger of the extrinsic pathway is the binding of pro-apoptotic ligands to cognate death receptors on the cell surface. This binding of ligands forms a death-inducing signaling complex (DISC), leading to activation of caspases 8 and 10. This process requires the death domain at the intracellular site of the death receptor [22]. Activated caspases 8 and 10 cleave effector caspases to amplify the death signal and then activate effector caspases 3, 6, and 7, which serve essential roles in both the intrinsic and extrinsic pathways as executioner caspases.
TRAIL cell surface receptors, which trigger the extrinsic pathway, are highly upregulated in a wide range of solid tumors. Therefore, TRAIL has been identified as an attractive therapeutic molecule for mediating apoptosis in tumor cells while sparing normal cells [23–25]. Activated death receptor 4 (DR4) and death receptor 5 (DR5) induce crosslinking scaffolds as well as tumor-associated leukocytes, which can further induce antibody-dependent, death receptor–mediated apoptosis in cancer cells while having no adverse effect on the proliferation of human T cells [26–28]. Additionally, these antibodies can stimulate NF-κB via a more distant receptor, CD40 [29].
TRAIL-R1 (DR4) agonistic antibodies include mapatumumab (HGS-ETR1) and AY4. AY4 was tested mainly in preclinical studies of anaplastic thyroid cancer and head and neck cancer and was found to induce reactive oxygen species–mediated apoptosis [30, 31]. Mapatumumab was tested in non–small cell lung cancer and showed a safe toxicity profile in preclinical studies. Unfortunately, neither agent demonstrated clinical efficacy [32, 33]. In triple-negative breast cancer (TNBC), TRAIL activation induced apoptosis in vitro, especially in cell lines with a mesenchymal phenotype, via DR5 or TRAIL receptor 2 (TRAIL-R2) in particular, or as well as via agonist antibodies mimicking the activity of TRAIL [34–36]. DR5-specific agonistic therapeutic antibodies include lexatumumab (HGS-ETR2), drozitumab, conatumumab (AMG-655), LBY 135, and tigatuzumab (CS-1008 or humanized TRA-8). Tigatuzumab was developed as a TRAIL humanized agonistic monoclonal antibody directed against DR5 [37, 38]. While the preclinical studies showed strong efficacy of tigatuzumab against TNBC/basal-like cells in vitro and in vivo when given in combination with paclitaxel and nab-paclitaxel [39–41], a phase II trial of tigatuzumab in combination with nab-paclitaxel resulted in only moderate prolongation of progression-free survival compared to nab-paclitaxel treatment alone for TNBC patients [42].
The death receptors are also involved in the extrinsic apoptosis pathway via epithelial–mesenchymal transition particularly E-cadherin [43]: loss of E-cadherin attenuates apoptotic signaling via DR4 and DR5, and the engagement of E-cadherin augments the activation of DR4 and DR5, which enhances the resulting progression of apoptosis. Most interestingly, E-cadherin boosts extrinsic apoptosis pathway signaling by coupling DR4 or DR5 to the actin cytoskeleton modulation.
The most exciting recent development in targeting the extrinsic pathway is the new-generation molecule ONC201 [44]. ONC201, an oral medication initially discovered as a TRAIL activity–inducing compound by drug sensitivity screening of the National Cancer Institute Library, induces selective apoptosis in cancer cells but is safe in normal tissue, acting via FOXO3a-mediated induction of the TRAIL gene and suppression of pAkt and pErk [45]. Further investigation of this molecule revealed that it induces an endoplasmic reticulum stress response in the cancer cells and induces binding to neurotransmitter receptors, including dopamine receptors. Most recently, the mechanism of action by which ONC201 induces apoptosis was shown to be inhibition of caseinolytic protease P [46••], a protease located in the inner mitochondrial membrane [47, 48]. Therefore, ONC201 also modulates the mitochondrial recruitment of the apoptosome through mitochondrial interaction—contributing to both extrinsic and intrinsic pathways. Clinical trials using extrinsic pathway targeted agents are summarized in Table 1.
Intrinsic Pathway and Targeted Agents
The cell-intrinsic apoptosis pathway is also known as the mitochondrial pathway; its signaling involves changes in the mitochondrial membranes and the release of proteins that result in widespread proteolysis and DNA cleavage [10]. This pathway is responsive to various genotoxic stresses, including conventional chemotherapeutics, radiation, and biologic agents that target cell survival and growth. The pro-apoptotic Bcl-2 family of proteins plays a critical role in this pathway, and the p53 tumor suppressor protein activates several of these pro-apoptotic family members. The intrinsic pathway has even been physically localized to the mitochondria [49]. The recent development of apoptosis-targeted agents focuses on these mitochondrial pathways and includes IAP inhibitors, Mcl-1 inhibitors, and Bcl-2 inhibitors.
IAP Inhibitors
Chemotherapy induces apoptosis in cancer cells more prominently compared to normal cells [50]; however, resistance develops by upregulation of inhibitors of apoptosis. Inhibitor of apoptosis proteins (IAPs) are an evolutionarily conserved family of proteins that are key negative regulators of both the intrinsic and extrinsic apoptotic pathways [51]. Proteins within this family include cIAP1, cIAP2, XIAP, NIAP, and survivin. In human cancer cell lines and tissues, one or more of these IAPs is overexpressed and inhibits apoptosis induction by targeting both intrinsic (mitochondrial) and extrinsic (death receptor) pathways of apoptosis [52].
IAP family members interfere with the induction of TNFR-mediated pro-immune responses through interleukin-mediated and NF-κB-mediated pathways [53]. Therefore, the inhibition of IAP could synergize with a checkpoint inhibitor or radiation-induced apoptosis, thus mediating effective tumor cell killing [54]. Inhibitor of apoptosis protein (IAP) is a crucial molecule for preventing effective apoptosis. Additionally, cIAP proteins ubiquitinate RIPK1, which facilitates the formation of IKK complex and promotes canonical NF-kB signaling. Activation of the NF-kB signaling pathway leads to induction of target genes that inhibit apoptosis (XIAP, Bcl-2, Bcl-xL) [55]. Therefore, therapeutics to inhibit the IAP molecule have been actively developed. The binding pockets of Smac and IAP share significant similarities [56]. Thus, many IAP inhibitors mimic the Smac protein as well (Smac mimetics).
LCL161 is the IAP inhibitor/Smac mimetic furthest along in the clinic [57, 58]. In hematological, colorectal, lung, and breast cancers [59, 60], LCL161 was tested as a single agent, and in combinations, and the clinical efficacies were mixed. In patients with myelofibrosis who progressed on JAK2 inhibitor, LCL161 once a week regimen has shown a safe toxicity profile and ability to maintain stable disease, therefore suggesting it as a potential future treatment option [61]. Given the mechanism of action, combination therapy with radiation has been tested in head and neck cancer and esophageal cancers. In preclinical studies in these cancers, LCL161 induced synergistic sensitivity to the radiation therapy; thereby, it was given in combination with radiation [62, 63]. The clinical outcomes are to be seen.
Other IAP inhibitors have been studied more in solid tumors. Birinapant is a novel bivalent small-molecule peptidomimetic of SMAC, shown to preferentially target cIAP1, cIAP2, and XIAP by binding to the BIR domains and trigger degradation through rapid RING-dependent autoubiquitylation [64, 65]. Inhibition of cIAP1/2 and XIAP with birinapant induces apoptosis through both the intrinsic and extrinsic pathways as well as through the canonical NF-kB pathway and can sensitize cancer cells to various apoptotic stimuli including radiation [54] and cytotoxic chemotherapy [66, 67].
In early phase clinical trials, birinapant has demonstrated tolerability and safety at effective doses, with a prolonged plasma half-life of 31 h and tumor half-life of 52 h. On target effect is the suppression of cIAP1, and it increases apoptosis in peripheral blood mononuclear cells and tumor tissue [68]. In a 5-arm phase I/II dose escalation study of birinapant administered intravenously in combination with different chemotherapies (docetaxel, irinotecan, gemcitabine, carboplatin/paclitaxel, liposomal doxorubicin) in patients with solid tumors, safety and tolerability were confirmed, and a phase II dose was established [69]. Interestingly, birinapant demonstrated prolonged progression-free survival in previously relapsed or refractory patients when combined with chemotherapies that induce TNFα, such as irinotecan [69]. While exciting results in preclinical models and early-phase trials have been seen, the clinical development of birinapant is currently on hold for unclear reasons. New IAP inhibitors such as ASTX660 are currently being tested in early phase clinical trials [70].
Survivin targeted antisense oligonucleotides (YM155, LY2181308) have been tested to induce the sensitization of cancer cells to therapeutics, yet the clinical efficacy has not been promising [71–74]. In mechanistic study, antisense YM155, despite early promising preclinical results, revealed an interference in DNA double-stranded break repair and topoisomerase-mediated DNA cleavage, which explains negative clinical efficacy. However, with rapid development of vaccine and medical chemistry technologies, the vaccine against survivin (SurVaxM, DPX-Survivac) [74] is showing the potential to be combined with immune checkpoint inhibitor therapies [2]. Currently ongoing clinical trials utilizing IAP inhibitors are summarized in Table 75.
Mcl-1 Inhibitors
An anti-apoptotic member of the Bcl-2 family, myeloid cell leukemia-1 (Mcl-1), was recently identified as a crucial apoptotic survival factor modulated by Wnt signaling in TNBC cells [76, 77]. Accumulated preclinical in vitro and in vivo evidence suggests that Mcl-1 represents a promising target for treating breast cancers [78–80]. Indeed, Mcl-1 is commonly amplified in 56% of TNBC tumors, and its overexpression is associated with high tumor grade and poor clinical prognosis [81, 82]. Furthermore, overexpression of Mcl-1 is implicated as a factor in resistance to multiple early- and advanced stage breast cancer therapies, such as microtubule-targeted agents paclitaxel and vincristine and the Bcl-2-targeting compound navitoclax [83–85]. Therefore, inhibition of Mcl-1 by targeted inhibitors represents an integrated approach for developing TNBC therapies by potentially restoring apoptotic signaling and rescuing the sensitivity to chemotherapy in Mcl-1-dependent TNBC tumors.
Currently under development in preclinical studies, Mcl-1 inhibitors have demonstrated great promise for the treatment of cancer, including breast cancer, in vitro and in vivo, as there now exist direct, potent, and selective Mcl-1 inhibitors with clear and specific cellular activity, disrupting Mcl-1 interactions and triggering apoptosis [86–88]. Moreover, the histone deacetylase inhibitor entinostat, together with the MEK inhibitor, pimasertib, has shown to mediate the targeted degradation of Mcl-1 through the induced expression of NOXA [89], a crucial regulator that fine-tunes cell death by targeting Mcl-1 for proteasomal degradation enhancing TNBC tumor cell death in vitro and in vivo [90, 91].
The targeted downregulation of Mcl-1 has been implicated in a phase I clinical trial to be the primary mechanism of activity of alvocidib (flavopiridol), a pan-CDK inhibitor, in patients with chronic lymphocytic lymphoma (CLL) or acute myeloid leukemia (AML) [92, 93]. While alvocidib is not a direct inhibitor of Mcl-1, these results have provided a rationale for an upcoming randomized phase II biomarker-driven clinical trial of alvocidib in patients with AML (NCT02520011), which could shed light on the implementation of Mcl-1 inhibition treatment for cancers. Several Mcl-1 inhibitors (AMG-176, AZD599) and CDK9 inhibitor that had shown effective suppression of Mcl-1 in preclinical studies are currently being tested in hematological malignancies (Table 3), and results are awaited.
Bcl-2 Inhibitors
BCL-2 family inhibitors are the front runners of the apoptosis-targeted agents developed in cancers. Each of these agents inhibits different family members such as BCL2, BCL-XL, and BCL-w, with different affinity [94, 95].
Venetoclax is the first agent that was approved by the FDA in cancer. It binds to BCL-2 protein and thereby displaces pro-apoptotic proteins like BIM and NOXA. This agent is approved in CLL, after showing a close to remarkable response even as a single agent [96]. Even in CLL with17p deletion, venetoclax showed similar response [96]. This is impressive given the aggressive behavior of 17 deletion CLL. When venetoclax was combined with rituximab, the response rate went up even higher up to 86% [97]. Venetoclax also showed significant activity in chemotherapy-resistant AML. As in CLL, TP53 mutation did not seem to reduce the efficacy of the agent, leading to the breakthrough approval by the FDA.
In breast cancer, venetoclax has been combined with fulvestrant to treat endocrine therapy–resistant, hormone receptor–positive breast cancer [98], given that functional estrogen receptor transcriptionally upregulates Bcl-2 as one of its direct target gene/proteins. Impressive results of combined tamoxifen and venetoclax sparked significant interest in combining venetoclax with various agents in breast cancer treatment [99•]. Unfortunately, the phase II trial VERONICA, testing the combination of venetoclax and fulvestrant, did not show any clinical efficacy [100].
Similarly, obatoclax mesylate (GX15-070) is another Bcl-2 inhibitor that has been tested in numerous cancers. Unfortunately, the toxicity profile of obatoclax included neurological (ataxia) symptoms and cytopenia which stopped obatoclax from further clinical development [101].
Navitoclax is another inhibitor in this category of agents. Navitoclax inhibits both Bcl-2 and Bcl-xL, thereby suppressing the compensatory emergence of Bcl-xL prevented by the release of pro-apoptotic regulators, like Bim [102, 4]. Bim can be replaced by inhibition of Bcl-2, but it can bind to other proteins of this category. Therefore, the inhibition of other Bcl-2 family proteins can synergistically induce Bim-mediated induction of apoptosis. While navitoclax, as a single agent, may not have shown remarkable clinical activity, combination with venetoclax has shown potential synergy [103•]. Clinical trials using venetoclax and navitoclax are summarized in Table 104.
Apoptosis and p53 Regulation
Cells with mutated or inactivated p53 develop resistance to apoptosis. As a compensatory mechanism, the p53 family member p73 can also inhibit apoptosis [105]. The essential pro-apoptotic genes induced by activated p53 within the cell-intrinsic apoptotic pathway include PUMA, NOXA, BAX, and Apaf-1. Both p53 and the FOXO family of transcription factors play an essential role in apoptosis by inducing the production of death receptors and pro-apoptotic Bcl-2 family proteins, thereby impacting both the intrinsic and extrinsic cell death pathways [106].
p53, the “guardian of the genome,” is an important regulator of apoptosis and other key biological functions [107]. The mouse double minute 2 (Mdm2) gene encodes a nuclear-localized E3 ubiquitin ligase, and its overexpression is detected in a variety of malignancies [108]. MDM2 binding to p53 induces p53 proteasomal degradation and inhibits p53 activity in apoptosis [109]. MDM2 inhibitor molecules can antagonize the p53-MDM2 interaction, allowing p53 to induce apoptotic pathways; hence, numerous preclinical and clinical studies have tested the efficacy of MDM2 inhibition. No active clinical developments to date have indicated unacceptable toxicity or suboptimal efficacy; however, clinical trials testing the efficacy of MDM2 inhibitors are ongoing, and the results are to be seen.
Kinase Inhibitor Targeting Apoptosis
MELK is a serine/threonine kinase in the AMPK family of kinases known to regulate cellular metabolism [110–110], regulate early embryonic development [110], and show elevated expression in human cancers [111, 112, 112–112]. It is an important proliferative marker and included as one of the genes included in MammaPrint [114] and PAM50 [115], both genomic assays used in breast cancer. High MELK expression is associated with poor overall and metastasis-free survival in many cancers [116, 121], including glioma cells [122, 123, 124]. Aside from contributions to several pro-cancer activities, MELK also regulates the activation of apoptosis [125].
Death-associated protein kinase (DAPK) is a serine/threonine kinase that comprises five family members (DAPK1-3, DRAK1 and DRAK2). This family of proteins has calcium/calmodulin domain and was previously known to be involved in important biological regulations including infection and neurosynaptic regulation which has been suggested as a promising target of Alzheimer disease treatment [126]. Recently, DAPK1 and DRAK2 have been shown to regulate autophagy, as well as apoptosis, contributing to metastatic progression (127). Small-molecule inhibitors against these proteins are under development, and the activity against cancers is to be seen.
Conclusions
Many cancer treatments rely on induction of effective apoptosis; hence, defects in apoptosis can render treatments ineffective. Despite the challenges of targeted drug development, companion biomarker development, and identification of appropriate groups of patient, targeting apoptosis remains a relevant strategy. We anticipate this field will continue to advance. After all, immunotherapy took more than 40 years to reach its “prime time.” With the right efforts and initiatives, we hope that apoptosis targeting opens up a new way to treat cancer. Combination of available agents and other therapeutics like radiation and immune checkpoint inhibitors also needs to be further explored and developed.
Data Availability
Not applicable
Code Availability
Not applicable
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science (New York, NY). 1997;277(5327):818–21.
Marsters SA, Pitti RA, Sheridan JP, Ashkenazi A. Control of apoptosis signaling by Apo2 ligand. Recent Prog Horm Res. 1999;54:225–34.
Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol: official journal of the American Society of Clinical Oncology. 1999;17(9):2941–53.
Kapur A, Felder M, Fass L, Kaur J, Czarnecki A, Rathi K, et al. Modulation of oxidative stress and subsequent induction of apoptosis and endoplasmic reticulum stress allows citral to decrease cancer cell proliferation. Sci Rep. 2016;6:27530.
Larsen BD, Sorensen CS. The caspase-activated DNase: apoptosis and beyond. FEBS J. 2017;284(8):1160–70.
Criscitiello C, Azim HA Jr, Schouten PC, Linn SC, Sotiriou C. Understanding the biology of triple-negative breast cancer. Ann Oncol. 2012;23(Suppl 6):vi13-8.
Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–18.
Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7(12):683–92.
King KL, Cidlowski JA. Cell cycle regulation and apoptosis. Annu Rev Physiol. 1998;60:601–17.
Lovric MM, Hawkins CJ. TRAIL treatment provokes mutations in surviving cells. Oncogene. 2010;29(36):5048–60.
Stadel D, Mohr A, Ref C, MacFarlane M, Zhou S, Humphreys R, et al. TRAIL-induced apoptosis is preferentially mediated via TRAIL receptor 1 in pancreatic carcinoma cells and profoundly enhanced by XIAP inhibitors. Clin Cancer Res. 2010;16(23):5734–49.
Choe SC, Hamacher-Brady A, Brady NR. Autophagy capacity and sub-mitochondrial heterogeneity shape Bnip3-induced mitophagy regulation of apoptosis. Cell Commun Signal. 2015;13:37.
Wolff S, Erster S, Palacios G, Moll UM. p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 2008;18(7):733–44.
Ramakrishnan V, Gomez M, Prasad V, Kimlinger T, Painuly U, Mukhopadhyay B, et al. Smac mimetic LCL161 overcomes protective ER stress induced by obatoclax, synergistically causing cell death in multiple myeloma. Oncotarget. 2016;7(35):56253–65.
Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–52.
Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42.
Pierceall WE, Kornblau SM, Carlson NE, Huang X, Blake N, Lena R, et al. BH3 profiling discriminates response to cytarabine-based treatment of acute myelogenous leukemia. Mol Cancer Ther. 2013;12(12):2940–9.
Zhang Z, Yang H, Wu G, Li Z, Song T, Li XQ. Probing the difference between BH3 groove of Mcl-1 and Bcl-2 protein: implications for dual inhibitors design. Eur J Med Chem. 2011;46(9):3909–16.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17(6):1675–87.
Ozoren N, El-Deiry WS. Defining characteristics of types I and II apoptotic cells in response to TRAIL. Neoplasia (New York, NY). 2002;4(6):551–7.
Roy S, Nicholson DW. Cross-talk in cell death signaling. J Exp Med. 2000;192(8):21–6.
Sessler T, Healy S, Samali A, Szegezdi E. Structural determinants of DISC function: new insights into death receptor-mediated apoptosis signalling. Pharmacol Ther. 2013;140(2):186–99.
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–82.
Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22(53):8628–33.
Rowinsky EK. Targeted induction of apoptosis in cancer management: the emerging role of tumor necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2005;23(36):9394–407.
Piechocki MP, Wu GS, Jones RF, Jacob JB, Gibson H, Ethier SP, et al. Induction of proapoptotic antibodies to triple-negative breast cancer by vaccination with TRAIL death receptor DR5 DNA. Int J Cancer. 2012;131(11):2562–72.
Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199(4):437–48.
Chattergoon MA, Muthumani K, Tamura Y, Ramanathan M, Shames JP, Saulino V, et al. DR5 activation of caspase-8 induces DC maturation and immune enhancement in vivo. Mol Ther. 2008;16(2):419–26.
Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19(1):101–13.
Lee BS, Kang SU, Hwang HS, Kim YS, Sung ES, Shin YS, et al. An agonistic antibody to human death receptor 4 induces apoptotic cell death in head and neck cancer cells through mitochondrial ROS generation. Cancer Lett. 2012;322(1):45–57.
Lee B, Cha H, Shin Y, Kim Y, Kim C. AY4, an agonistic anti-death receptor 4 MAB, induces apoptotic cell death in anaplastic thyroid cancer cells via downregulation of Bcl-xL with reactive oxygen species generation. Endocrine-related cancer. 2013;20(3).
Greco FA, Bonomi P, Crawford J, Kelly K, Oh Y, Halpern W, et al. Phase 2 study of mapatumumab, a fully human agonistic monoclonal antibody which targets and activates the TRAIL receptor-1, in patients with advanced non-small cell lung cancer. Lung cancer (Amsterdam, Netherlands). 2008;61(1):82–90.
Mom CH, Verweij J, Oldenhuis CNAM. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res. 2009;15(17):5584–90.
Rahman M, Davis SR, Pumphrey JG, Bao J, Nau MM, Meltzer PS, et al. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat. 2009;113(2):217–30.
Shi J, Zheng D, Liu Y, Sham MH, Tam P, Farzaneh F, et al. Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res. 2005;65(5):1687–92.
Clancy L, Mruk K, Archer K, Woelfel M, Mongkolsapaya J, Screaton G, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA. 2005;102(50):18099–104.
Yada A, Yazawa M, Ishida S, Yoshida H, Ichikawa K, Kurakata S, et al. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann Oncol. 2008;19(6):1060–7.
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7(8):954–60.
Buchsbaum DJ, Zhou T, Grizzle WE, Oliver PG, Hammond CJ, Zhang S, et al. Antitumor efficacy of TRA-8 anti-DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin Cancer Res. 2003;9(10 Pt 1):3731–41.
Oliver PG, LoBuglio AF, Zhou T, Forero A, Kim H, Zinn KR, et al. Effect of anti-DR5 and chemotherapy on basal-like breast cancer. Breast Cancer Res Treat. 2012;133(2):417–26.
Londono-Joshi AI, Oliver PG, Li Y, Lee CH, Forero-Torres A, LoBuglio AF, et al. Basal-like breast cancer stem cells are sensitive to anti-DR5 mediated cytotoxicity. Breast Cancer Res Treat. 2012;133(2):437–45.
Forero-Torres A, Varley KE, Abramson VG, Li Y, Vaklavas C, Lin NU, et al. TBCRC 019: a phase II trial of nanoparticle albumin-bound paclitaxel with or without the anti-death receptor 5 monoclonal antibody tigatuzumab in patients with triple-negative breast cancer. Clin Cancer Res. 2015;21(12):2722–9.
Lu M, Marsters S, Ye X, Luis E, Gonzalez L, Ashkenazi A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Molecular cell. 2014;54(6).
Allen JE, Kline CLB, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7(45):74380–92.
Allen J, Krigsfeld G, Mayes P, Patel L, Dicker D, Patel A, et al. Dual inactivation of Akt and ERK by TIC10 Signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5(171):171ra17.
•• Ishizawa J, Zarabi S, Davis R, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer cell. 2019;35(5). (This study found a direct mechanism of action of one of the most promising extrinsic apoptosis inducing agent ONC201, potentially opening a new avenue of apoptosis-targeted cancer therapeutics.)
Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17.
Wagner J, Kline CL, Zhou L, Campbell KS, MacFarlane AW, Olszanski AJ, et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest. 2018;128(6):2325–38.
Cheng E, Wei M, Weiler S, Flavell R, Mak T, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and. Mol Cell. 2001;8(3):705–11.
Green DR. Cancer and apoptosis: Who Is Built to Last? Cancer Cell. 2017;31(1):2–4.
LaCasse E, Baird S, Korneluk R, MacKenzie A. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1999;17(25).
Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ, Kaufmann SH. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J Biol Chem. 2002;277(46):44236–43.
Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9(12):1371–8.
Cerna D, Lim B, Adelabu Y, Yoo S, Carter D, Fahim A, et al. SMAC mimetic/IAP inhibitor birinapant enhances radiosensitivity of glioblastoma multiforme. Radiat Res. 2021;195(6):549–60.
Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12(7):439–52.
Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J, et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature. 1999;401(6755):818–22.
Pemmaraju N, Carter B, Kantarjian H, Cortes J, Kadia T, Garcia-Manero GD, CD, et al. Results for phase II clinical trial of LCL161, a SMAC mimetic, in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (post-PV MF) or post-essential thrombocytosis myelofibrosis (post-ET MF). ASH 58th Annual Meeting & Exposition Proceedings. 2016;Blood 2016 128:3105.
Infante J, Dees E, Olszanski A, Dhuria S, Sen S, Cameron S, et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2014.
Gerges S, Rohde K, Fulda S. Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett. 2016;375(1):127–32.
L Vidal R, Dees E, Chia S. A phase Ib study of LCL161, an oral inhibitor of apoptosis (IAP) antagonist, in combination with weekly paclitaxel in patients with advanced solid tumors. Cancer Research. 2012;72(24, Suppl 3).
Pemmaraju N, Carter BZ, Kantarjian HM, Cortes JE, Bose P, Kadia TM, et al. Final results of phase 2 clinical trial of LCL161, a novel oral SMAC mimetic/IAP antagonist, for patients with intermediate to high risk myelofibrosis. Blood. 2019;134(Supplement_1):555.
Qin Q, Zuo Y, Yang X, Lu J, Zhan L, L X, et al. Smac mimetic compound LCL161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(3).
Yang L, Kumar B, Shen C, Zhao S, Blakaj D, Li T, et al. LCL161, a SMAC-mimetic, preferentially radiosensitizes human papillomavirus-negative head and neck squamous cell carcinoma. Molecular cancer therapeutics. 2019;18(6).
Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57(9):3666–77.
Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Mol Cancer Ther. 2014;13(4):867–79.
Carter BZ, Mak PY, Mak DH, Shi Y, Qiu Y, Bogenberger JM, et al. Synergistic targeting of AML stem/progenitor cells with IAP antagonist birinapant and demethylating agents. J Natl Cancer Inst. 2014;106(2):djt440.
Min DJ, He S, Green JE. Birinapant (TL32711) Improves responses to GEM/AZD7762 combination therapy in triple-negative breast cancer cell lines. Anticancer Res. 2016;36(6):2649–57.
Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, et al. A phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther. 2015;14(11):2569–75.
Amaravadi RK, Senzer NN, Martin LP, Schilder RJ, LoRusso P, Papadopoulos KP, et al. A phase I study of birinapant (TL32711) combined with multiple chemotherapies evaluating tolerability and clinical activity for solid tumor patients. J Clin Oncol. 2013;31(15_suppl):2504.
Ward GA, Lewis EJ, Ahn JS, Johnson CN, Lyons JF, Martins V, et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNFalpha-dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol Cancer Ther. 2018;17(7):1381–91.
Wiechno P, Somer BG, Mellado B, Chlosta PL, Cervera Grau JM, Castellano D, et al. A randomised phase 2 study combining LY2181308 sodium (survivin antisense oligonucleotide) with first-line docetaxel/prednisone in patients with castration-resistant prostate cancer. Eur Urol. 2014;65(3):516–20.
Natale R, Blackhall F, Kowalski D, Ramlau R, Bepler G, Grossi F, et al. Evaluation of antitumor activity using change in tumor size of the survivin antisense oligonucleotide LY2181308 in combination with docetaxel for second-line treatment of patients with non-small-cell lung cancer: a randomized open-label phase II study. J Thorac Oncol: official publication of the International Association for the study of lung cancer. 2014;9(11):1704–8.
Yu Y, Zhao X, Zhang Y, Kang Y, Wang J, Liu Y. Antitumor activity of YM155, a selective survivin suppressant, in combination with cisplatin in hepatoblastoma. Oncol Rep. 2015;34(1):407–14.
Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother CII. 2016;65(11):1339–52.
Dorigo O, Fiset S, MacDonald L, Bramhecha Y, Hrytsenko O, Dirk B, et al. DPX-Survivac, a novel T-cell immunotherapy, to induce robust T-cell responses in advanced ovarian cancer. https://doi.org/10.1200/JCO.2020.38.5_suppl.6
Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015.
Yang L, Perez AA, Fujie S, Warden C, Li J, Wang Y, et al. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer. 2014;14:124.
Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24(2):182–96.
Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012;21(4):547–62.
Liu X, Tang H, Chen J, Song C, Yang L, Liu P, et al. MicroRNA-101 inhibits cell progression and increases paclitaxel sensitivity by suppressing MCL-1 expression in human triple-negative breast cancer. Oncotarget. 2015;6(24):20070–83.
Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–45.
Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67(10):4564–71.
Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–4.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–99.
Boiani M, Daniel C, Liu X, Hogarty MD, Marnett LJ. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. J Biol Chem. 2013;288(10):6980–90.
Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13(3):565–75.
Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015;6:e1590.
Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P, et al. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther. 2010;10(9):903–17.
Torres-Adorno AM, Lee J, Kogawa T, Ordentlich P, Tripathy D, Lim B, et al. Histone deacetylase inhibitor enhances the efficacy of MEK inhibitor through NOXA-mediated MCL1 degradation in triple-negative and inflammatory breast cancer. Clin Cancer Res. 2017;23(16):4780–92.
Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(Suppl 1):S84-92.
Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26(4):778–87.
Dettman EW, SL; Doykan, C; Arn, M; Blake, N; Bearss, DJ; Cardone, M; Smith, BD editor mitochondrial profiling in AML patients treated with an alvocidib containing regimen reveals MCL1 dependency in responder bone marrow. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 2015 Apr 18–22; Philadelphia, PA: AACR.
Whatcott C, editor The MCL-1 targeting effect of alvocidib potentiates the activity of cytarabine and mitoxantrone in a time-sequential regimen in AML. SOHO 2015 Annual Meeting; 2015 2015, Sept-16; Houston, TX.
Pecot J, Maillet L, Le Pen J, Vuillier C, Trecesson SC, Fetiveau A, et al. Tight sequestration of BH3 proteins by BCL-xL at subcellular membranes contributes to apoptotic resistance. Cell Rep. 2016;17(12):3347–58.
Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7(279):279ra40.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–22.
Seymour JF, Ma S, Brander DM, Choi MY, Barrientos J, Davids MS, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18(2):230–40.
Elledge RM, Green S, Howes L, Clark GM, Berardo M, Allred DC, et al. bcl-2, p53, and response to tamoxifen in estrogen receptor-positive metastatic breast cancer: a Southwest Oncology Group study. J Clin Oncol: official journal of the American Society of Clinical Oncology. 1997;15(5):1916–22.
• Lindeman G, Hamilton E, Krop I, Lim B, Modi S, Saura C, et al. Abstract OT-28–03: VICKI: a phase Ib/II, randomized, placebo-controlled, study of venetoclax plus ado-trastuzumab emtansine (T-DM1) in patients (pts) with previously treated HER2-positive locally advanced (LA) or metastatic breast cancer (MBC). 2021;81(4_suppl). (One of the first studies in solid tumor showing the potential synergy of Bcl-2 inhibitor and standard anti-Her2 therapy. This study provided excitement and rationale to investigate Bcl-2 inhibitors in solid tumors.)
Lindeman GJ, Bowen R, Jerzak KJ, Song X, Decker T, Boyle FM, et al. Results from VERONICA: a randomized, phase II study of second-/third-line venetoclax (VEN) + fulvestrant (F) versus F alone in estrogen receptor (ER)-positive, HER2-negative, locally advanced, or metastatic breast cancer (LA/MBC). https://doi.org/10.1200/JCO.2021.39.15_suppl.1004. 2021;39(15_suppl.).
Goy A, Hernandez-Ilzaliturri FJ, Kahl B, Ford P, Protomastro E, Berger M. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma. 2014;55(12):2761–8.
Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med. 2017;9(420).
Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT, et al. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood. 2011;117(26):7145–54.
• Pullarkat V, Lacayo N, Jabbour E, Rubnitz J, Bajel A, Laetsch T, et al. Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer discovery. 2021;11(6). (A study showing the clinical efficacy of combined Bcl-2 inhibitor and Bcl-xL inhibitor in hematological malignancy opened a case for potential synergy by combining inhibitors of the same BH3 domain sharing Bcl-2 family proteins.)
Gong J, Costanzo A, Yang H-Q, Melino G, Kaelin WG, Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–9.
Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochem Biophys Acta. 2011;1813(11):1978–86.
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–6.
Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb Perspect Med. 2016;6(6):a026336.
Trino S, De Luca L, Laurenzana I, Caivano A, Del Vecchio L, Martinelli G, et al. P53-MDM2 pathway: evidences for a new targeted therapeutic approach in B-acute lymphoblastic leukemia. Front Pharmacol. 2016;7:491.
Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31(6):1051–63.
Li S, Li Z, Guo T, Xing XF, Cheng X, Du H, et al. Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer. Oncotarget. 2016;7(5):6266–80.
Wang Y, Lee YM, Baitsch L, Huang A, Xiang Y, Tong H, et al. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife. 2014;3:e01763.
Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23(4):833–43.
Sun X, Gao L, Chien HY, Li WC, Zhao J. The regulation and function of the NUAK family. J Mol Endocrinol. 2013;51(2):R15-22.
Heyer BS, Warsowe J, Solter D, Knowles BB, Ackerman SL. New member of the Snf1/AMPK kinase family, Melk, is expressed in the mouse egg and preimplantation embryo. Mol Reprod Dev. 1997;47(2):148–56.
Speers C, Zhao SG, Kothari V, Santola A, Liu M, Wilder-Romans K, et al. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin Cancer Res. 2016;22(23):5864–75.
Du T, Qu Y, Li J, Li H, Su L, Zhou Q, et al. Maternal embryonic leucine zipper kinase enhances gastric cancer progression via the FAK/Paxillin pathway. Mol Cancer. 2014;13:100.
Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin Transl Med. 2015;4:11.
Inoue H, Kato T, Olugbile S, Tamura K, Chung S, Miyamoto T, et al. Effective growth-suppressive activity of maternal embryonic leucine-zipper kinase (MELK) inhibitor against small cell lung cancer. Oncotarget. 2016;7(12):13621–33.
Kato T, Inoue H, Imoto S, Tamada Y, Miyamoto T, Matsuo Y, et al. Oncogenic roles of TOPK and MELK, and effective growth suppression by small molecular inhibitors in kidney cancer cells. Oncotarget. 2016;7(14):17652–64.
Pickard MR, Green AR, Ellis IO, Caldas C, Hedge VL, Mourtada-Maarabouni M, et al. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res: BCR. 2009;11(4):R60.
Tian S, Roepman P, Van’t Veer LJ, Bernards R, de Snoo F, Glas AM. Biological functions of the genes in the mammaprint breast cancer profile reflect the hallmarks of cancer. Biomark Insights. 2010;5:129–38.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2009;27(8):1160–7.
Hebbard LW, Maurer J, Miller A, Lesperance J, Hassell J, Oshima RG, et al. Maternal embryonic leucine zipper kinase is upregulated and required in mammary tumor-initiating cells in vivo. Cancer Res. 2010;70(21):8863–73.
Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015;4(2):226–38.
Chen D, Zhou X, Lee T. Death-associated protein kinase 1 as a promising drug target in cancer and Alzheimer’s disease. Recent Pat Anti-Cancer Drug Discovery. 2019;14(2):144–57.
Wu YM, Chen ZJ, Jiang GM, Zhang KS, Liu Q, Liang SW, et al. Inverse agonist of estrogen-related receptor alpha suppresses the growth of triple negative breast cancer cells through ROS generation and interaction with multiple cell signaling pathways. Oncotarget. 2016;7(11):12568–81.
Acknowledgements
The manuscript was edited by Sarah Bronson, ELS, of the Research Medical Library at The University of Texas MD Anderson Cancer Center.
Author information
Authors and Affiliations
Contributions
BL and PS
Corresponding authors
Ethics declarations
Conflict of Interest
Puneet Singh declares that she has no conflict of interest. Bora Lim has received research funding from Puma Biotechnology, Novartis, Genentech, Merck, and Takeda Oncology. There are no directly relevant financial activities related to the drugs included in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical collection on Evolving Therapies
Rights and permissions
About this article

Cite this article
Singh, P., Lim, B. Targeting Apoptosis in Cancer. Curr Oncol Rep 24, 273–284 (2022). https://doi.org/10.1007/s11912-022-01199-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-022-01199-y