
Abstract
Purpose of Review
The upcoming 2021 World Health Organization (WHO) Classification of Tumours of the Central Nervous System will feature numerous changes in classification, diagnostic criteria, nomenclature, and grading of diffuse gliomas. This article reviews these changes and the clinical and molecular findings underlying them.
Recent Findings
Since the publication of the 2016 World Health Organization (WHO) Classification of Tumours of the Central Nervous System, research has led to new insights into how molecular changes impact both the classification and grading of CNS tumors. The continued integration of molecular and histopathological features has led to changes in diagnostic criteria and grading for various tumors. In the new 2021 WHO CNS tumor classification scheme, diffuse gliomas will be classified as either adult-type diffuse gliomas, pediatric-type diffuse high-grade gliomas, or pediatric-type diffuse low-grade gliomas.
Summary
The upcoming changes in the classification of adult-type and pediatric-type diffuse gliomas allow for more effective communication of both diagnostic and prognostic information, and—particularly in the case of pediatric-type diffuse gliomas—may suggest possible targeted strategies for therapeutic intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The explosion of cancer genomics over the past two decades has left an indelible impact on diagnostic pathology. Across the broad spectrum of disease, morphologically based classification systems are being significantly revised, if not overturned completely, in the face of compelling molecular signatures with demonstrable prognostic and/or predictive significance. Perhaps nowhere has this paradigm shift been more apparent than for neoplasms of the central nervous system (CNS), particularly diffusely infiltrating gliomas (aka diffuse gliomas), the most common primary brain tumors. As with other tumor types, diffuse gliomas had been morphologically classified on the basis of presumed histogenesis (see below). However, recently elucidated molecular features, spanning both genomic and epigenomic landscapes, have clearly delineated more well-defined and biologically uniform disease entities, whose constituencies transcend conventional histopathological designations. These developments have led to significant diagnostic revisions of the World Health Organization (WHO) Classification of Tumors of the Central Nervous System, initially evident in the 2016 revision of the 2007 edition (WHO 2016), and now even more tangibly present in the 2021 edition (WHO 2021).
Since WHO 2016, defining molecular features have been incorporated as essential elements in so-called “integrated diagnoses”, a trend that broadens further in WHO 2021, for diffuse gliomas as well as several other brain tumor classes. This fundamental shift in approach implicitly acknowledges that histopathologically similar or even indistinguishable tumors may possess different patterns of molecular alteration and, most importantly, distinct clinical outcomes.
Of note, in a break with tradition, the 2021 WHO CNS tumor classification shifts toward the use of Arabic numerals (e.g., 1, 2, 3, 4) instead of Roman numerals for the designation of tumor grade [1••]. Not only does this bring CNS tumor grading in line with WHO grading used in tumors involving other organ systems, but also reduce the risk of confusion due to the similarities in appearance between Roman numerals (e.g., “II” vs “III”)—errors that may propagate through the medical record, leading to unintended and potentially unfavorable clinical consequences [1••, 2]. We will use this new Arabic numeral convention moving forward in this manuscript unless deliberately referring to legacy nomenclature. WHO 2021 has also drawn stark distinctions between diffuse gliomas impacting primarily adults and those affecting primarily children. Three broad categories have emerged from this partitioning—adult-type diffuse gliomas, pediatric-type diffuse high-grade gliomas, and pediatric-type diffuse low-grade gliomas (Table 1). This review will address each of these tumor subsets in turn, focusing on the significant differences between WHO 2016 and WHO 2021.
Adult-Type Diffuse Gliomas
These tumors have historically been designated by morphological considerations into astrocytoma (WHO grades II and III), oligodendroglioma (WHO grade II and III), and glioblastoma (WHO grade IV). A mixed oligoastrocytoma category was eliminated in WHO 2016 due to an absence of defining molecular characteristics. In WHO 2021, this basic astrocytoma/oligodendroglioma/glioblastoma nomenclature remains, although the corresponding disease entities have been further refined and specified by the integration of specific biomarkers. Adult-type diffuse gliomas are broadly classified as either astrocytoma, IDH-mutant, oligodendroglioma, IDH-mutant and 1p/19q-codeleted, or glioblastoma, IDH-wild type.
IDH-Mutant Astrocytomas
The WHO 2016 recognized the fundamental molecular and clinical distinctions that exist between IDH-mutant and IDH-wild type diffuse astrocytomas, with the latter characterized by an intrinsically more aggressive clinical course [3]. This basic concept persists in WHO 2021, with formalized changes in nomenclature underscoring the profound differences in biological potential exhibited by these diagnostic categories. Specifically, while WHO 2016 employed identical terminology for both IDH-wildtype and IDH-mutant astrocytomas—diffuse astrocytoma (Grade II), anaplastic astrocytoma (Grade III), and glioblastoma (Grade IV)[3]—WHO 2021 designates IDH-mutant diffuse astrocytomas as either astrocytoma, IDH-mutant, WHO Grade 2, astrocytoma, IDH-mutant, WHO Grade 3, or astrocytoma, IDH-mutant, WHO Grade 4. Importantly, the term glioblastoma, IDH-mutant, WHO grade IV, introduced to WHO 2016 to denote the fully malignant state of IDH-mutant astrocytoma, has been discontinued and replaced with astrocytoma, IDH-mutant, WHO Grade 4 to emphasize the IDH-mutant vs IDH-wildtype pathogenic distinction. Analogous changes have been instituted for IDH-wild type glioblastoma (see below).
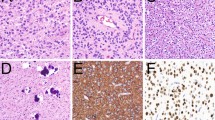
Although precise morphological features may vary, IDH-mutant astrocytomas are mildly to moderately hypercellular relative to normal brain and typically composed of fibrillary glial cells that diffusely infiltrate into surrounding parenchyma (Fig. 1a). Microcytic matrix formation in the background is a common feature. By definition, these tumors feature point mutations in IDH1 or IDH2, with the vast majority also harboring loss-of-function mutations involving TP53 and ATRX (Fig. 1b–d). This combination of molecular alterations greatly facilitates diagnosis through either immunohistochemical or focused molecular testing. As in WHO 2016, microvascular proliferation and/or necrosis designate grade 4 classification, while grades 2 and 3 are distinguished by the latter showing histologic features of anaplasia (at least focally) and “significant” mitotic activity. While no definitive criteria exist for what constitutes “’significant’ mitotic activity,” a standard of 2 or more mitoses in a resection specimen, or a single mitotic figure in minute biopsies is generally accepted [2, 3]. In recent studies, homozygous deletions of CDKN2A/B have been demonstrated to be strongly associated with shorter survival in patients diagnosed with IDH-mutant diffuse astrocytic gliomas regardless of grade [4•, 5•, 6•]. Accordingly, homozygous CDKN2A/B deletions are now included with microvascular proliferation and necrosis as definitive criteria designating WHO grade 4 status in this tumor group.

Astrocytoma, IDH-mutant. H&E staining reveals a moderately cellular neoplasm composed of infiltrating fibrillary neoplastic glial cells arranged in a vaguely microcystic background (a). The tumor cells express the mutant IDH1 R132H protein (b), along with p53 (c, arrowhead). Nuclear expression of ATRX is lost (d, arrowhead). 200 × magnification
Finally, very recent work has identified rare, infratentorial IDH-mutant gliomas as molecularly and clinically distinct disease variants [7]. Specifically, these tumors exhibit higher rates of “non-canonical” (non IDH1 R132H) mutations in IDH1 and IDH2, lower rates of ATRX deficiency, isolated co-occurrence of H3K27M mutation (see below), and relatively unfavorable clinical outcome compared to their much more common, supratentorial counterparts.
Oligodendroglioma, IDH-Mutant, and 1p/19q-Codeleted
Oligodendrogliomas consist of monomorphic tumor cells with round to oval nuclei (resembling oligodendrocytes) that characteristically exhibit perinuclear cytoplasmic clearing on formalin-fixed paraffin-embedded sections (Fig. 2a); microcalcifications as well as a network of fine arborizing vessels are common [3]. In contrast to IDH-mutant astrocytomas, oligodendrogliomas are defined by the combination of IDH1/IDH2 mutation and coincident loss of chromosomes 1p and 19q (1p/19q codeletion). This classification scheme was initially adopted in WHO 2016 and essentially remains unchanged for WHO 2021. WHO grade 3 (anaplastic) status is conferred on an IDH-mutant, 1p/19q codeleted oligodendroglioma that features dense cellularity, microvascular proliferation, necrosis, and/or “significant” mitotic activity, with 6 mitoses per 10 high-power fields in a resection specimen, or a single mitotic figure in minute biopsy generally accepted as sufficient [3] (Fig. 2b). That being said, formal thresholds for proliferative activity designating WHO grade 3 status have yet to be established.

Oligodendroglioma, IDH-mutant and 1p/19q codeleted. WHO grade 2 oligodendrogliomas are composed of infiltrating tumor cells with monomorphic round to oval nuclei and perinuclear halos (a). WHO grade 3 variants (b) exhibit more dense cellularity and proliferative activity and may contain microvascular proliferation (red arrowhead) and/or necrosis (green arrowhead). 200 × magnification
Glioblastoma, IDH-Wildtype
In WHO 2021, the entity of glioblastoma will be fully reconceived as an IDH-wild type diffuse astrocytoma by definition. In this molecular context, the morphological features classically associated with glioblastoma, namely microvascular proliferation (Fig. 3) and necrosis, will each continue to serve as diagnostic criteria. However, additional molecular factors, above and beyond the absence of IDH1/2 mutation, will also designate this diagnosis. Specifically, EGFR amplification, TERT promoter mutation, and combined whole chromosome 7 gain and whole chromosome 10 loss (+ 7/-10) have been extensively implicated by recent literature as defining alterations for IDH-wild type glioblastoma, even in the absence of microvascular proliferation or necrosis [1••, 4•, 8••]. Additional molecular alterations, such as CDK4 amplification, MDM2 amplification, MET amplification, PDGFRA amplification/mutation, and FGFR3 fusion, are also highly suggestive of IDH-wild type glioblastoma in the appropriate setting [9].

Glioblastoma, IDH-wild type. High-power field demonstrating classic glomeruloid microvascular proliferation (arrowhead). 400 × magnification
IDH-wildtype diffuse astrocytic gliomas that do not harbor either microvascular proliferation, necrosis, or the cardinal molecular alterations of glioblastoma (EGFR amplification, TERT promoter mutation, + 7/-10) will be designated either diffuse astrocytoma, IDH-wildtype (WHO Grade 2) or anaplastic astrocytoma, IDH-wildtype (WHO Grade 3), once again with “significant” mitotic activity conferring higher grade [3]. These latter diagnostic categories likely represent an amalgamation of multiple biological entities, whose precise composition may become apparent with additional molecular profiling. Indeed, some such tumors may reflect pediatric-type diffuse gliomas (see below) arising in adult patient populations.
Pediatric-Type Diffuse Gliomas
Despite the longstanding appreciation within the neuropathology community that diffuse gliomas primarily impacting children and young adults are likely distinct from their adult counterparts on a molecular level, conventional morphological classification (e.g., astrocytoma, oligodendroglioma, glioblastoma) has stubbornly persisted through repeated iterations of WHO schema. WHO 2021 dramatically departs from this practice, with a number of new diagnostic entities delineated by recent integrated molecular profiling studies.
WHO 2021 divides pediatric-type diffuse gliomas into two broad groups corresponding to high-grade and low-grade clinical behavior. The former includes diffuse midline glioma, H3 K27-altered, diffuse hemispheric glioma, H3 G34-mutant, diffuse pediatric-type high grade glioma, H3 wildtype and IDH-wildtype, and infant-type hemispheric glioma. The latter includes diffuse astrocytoma, MYB or MYBL1-altered, angiocentric glioma, polymorphous low-grade neuroepithelial tumor of the young, and diffuse low-grade glioma, MAPK pathway-altered.
Pediatric-Type Diffuse High-Grade Gliomas
The molecular distinctiveness of pediatric high-grade gliomas was emphatically confirmed with the discovery of histone H3 mutations almost 10 years ago [10, 11], a breakthrough that led directly to the designation of diffuse midline glioma, H3 K27M-mutant, WHO grade IV, as a novel tumor entity in WHO 2016. Over the past 5 years, additional genomic analyses have continued to refine conceptions of diffuse midline glioma, along with other biologically specified glioma variants affecting primarily, though not exclusively, children and young adults.
Diffuse Midline Glioma, H3 K27-Altered
These high-grade, infiltrative gliomas with astrocytic morphology (Fig. 4a) are defined by alterations involving the Lysine 27 residue of either H3F3A or HIST1H3B/C, with conversion to methionine (K27M) occurring in the vast majority cases [12•]. Initially included in WHO 2016 as diffuse midline glioma, H3 K27M-mutant, WHO grade IV, this tumor type expanded the previous notion of diffuse intrinsic pontine glioma, an aggressive brainstem neoplasm arising in children, to include similar lesions up and down the central neuraxis occurring across a more diverse age range, although still concentrated in the pediatric population [3]. The robustness of this diagnostic category has been repeatedly tested since its conception. In particular, multiple studies have now shown that the H3 K27M mutation, initially thought to be entirely restricted to diffuse midline gliomas with invariably poor prognoses, arises infrequently in other morphological brain tumor subtypes, some of which—like pilocytic astrocytoma and ganglioglioma—exhibit relatively favorable clinical behavior [13,14,15]. Such findings led the cIMPACT-NOW to recommend that in order to render the diagnosis of diffuse midline glioma, H3 K27M-mutant, the tumor in question must be (1) a glioma, (2) infiltrative, (3) midline, and (4) H3 K27M-mutated [12•].

Diffuse midline glioma, H3 K27-altered. H&E staining demonstrates an infiltrative astrocytic neoplasm (a). Immunostaining for H3 K27me3 is lost in tumor cell nuclei (b), with retained expression in background microvasculature (arrowhead). 200 × magnification
Recent work has also identified additional molecular alterations impacting this class of tumor, distinct from H3 K27M mutation, that nonetheless similarly impact H3 K27 functionality, specifically by inhibiting the extent to which histone K27 residues genome-wide undergo repressive trimethylation (H3 K27me3). These alterations include H3K27I mutation and overexpression of the EZHIP protein, the latter of which effectively serves as a peptide mimic of the K27M-mutated moiety [16, 17]. These exciting developments have prompted a further revision of this diagnostic entity to diffuse midline glioma, H3 K27-altered. Importantly, it should be recognized that immunostaining for the H3 K27M mutation may no longer capture all diffuse midline gliomas. Accordingly, diagnostic algorithms should also include the application of the more sensitive, though less specific H3 K27me3 immunostain, which should demonstrate loss of nuclear expression in all cases (Fig. 4b), even those without H3 K27M mutation [18].
Diffuse Hemispheric Glioma, H3 G34-Mutant
The initial discovery of histone H3 abnormalities in glioma also delineated a distinct set mutations involving the glycine 34 residue of H3F3A and rarely H3F3B, with conversion to either arginine or valine [1••, 10, 19]. It was soon appreciated that G34R/V-mutant pediatric high-grade gliomas localized entirely to the cerebral hemispheres, in sharp contrast to their K27M-mutant counterparts. Histologically, these tumors are diffuse infiltrating gliomas with astrocytic differentiation and anaplastic features, including elevated mitotic activity, microvascular proliferation, and necrosis (Fig. 5). Frankly, embryonal architecture with dense cellularity and high nuclear/cytoplasmic ratios can also be seen, and particularly in this setting, microvascular proliferation and necrosis may be lacking [1••]. In spite of this variable morphology, these tumors exhibit “uniform epigenetic signatures, suggesting a single biological origin” [20]. In this regard, recent work has implicated neuronal, rather than glial, precursors as cells-of-origin for G34R/V-mutant gliomas [21, 22]. Much like IDH-mutant astrocytomas in adults, ATRX and TP53 mutations are present in nearly all cases, with coincident loss of ATRX expression and p53 overexpression on immunostaining. Accordingly, ATRX immunohistochemistry provides a sensitive screening tool for the detection of both neoplastic variants. G34R/V-mutant high-grade glioma exhibits an aggressive clinical course corresponding to WHO grade 4, though prognosis is generally not as grim as for diffuse midline glioma.

Diffuse hemispheric glioma, H3 G34-mutant. Similar to glioblastomas in adults, these tumors frequently exhibit microvascular proliferation (red arrowhead) and pseudopalisading necrosis (green arrowhead). 200 × magnification
Diffuse Pediatric-Type High Grade Glioma, H3 Wildtype, and IDH-Wildtype
These tumors, arising in children and young adults, comprise most of the remaining high-grade diffuse gliomas that are both IDH-wildtype and histone H3-wildtype. Their molecular heterogeneity is underscored by the variety of genomic driver alterations implicated in their pathogenesis, including amplification/mutation of receptor tyrosine kinase genes like EGFR and PDGFRA, amplification of MYCN, mutation/alteration of TP53 and NF1, and TERT promoter mutation [23••]. As such, diagnostic overlap with genomically similar IDH-wild type adult glioblastomas is likely. High-grade gliomas arising in the context of constitutional mismatch repair deficiency (cMMRD), Lynch Syndrome or Li Fraumeni syndrome exhibit a similar spectrum of molecular alterations to those described here but should be considered distinct from spontaneously arising tumors within this diagnostic category [24].
Infant-Type Hemispheric Glioma
Generally arising in patients younger than 12 months of age, these gliomas are clinically and molecularly distinct from those affecting older children and adults [25]. Morphologically, infantile-type hemispheric gliomas tend to show uniform architecture and significant nuclear pleomorphism. Notably, histologically high-grade tumors may have better clinical outcomes compared to histologically low-grade tumors within the infant population [26]. Two-thirds of infant-type hemispheric glioma are RTK-driven and often harbor ALK, NTRK1/2/3, ROS1, or MET gene fusions [25, 26], potentially rendering them ideal candidates for targeted therapy. Another subset that lacks RTK fusions tends to have a worse prognosis, with no definitive recurrent genetic driver as yet been identified [25].
Pediatric-Type Diffuse Low-Grade Gliomas
Large-scale molecular profiling within this heterogeneous tumor group has led to the recent refinement of multiple entities with distinct biological and clinical features. Given the current pace of discovery, the delineation of additional subtypes in the near future is likely.
Diffuse Astrocytoma, MYB, or MYBL1-Altered; Diffuse Low-Grade Glioma, MAPK Pathway-Altered
These epilepsy-associated infiltrating gliomas exhibit astrocytic or oligodendroglial morphology, demonstrate no microvascular proliferation or necrosis, and have little to no proliferative activity [27]. The indolent biological behavior of these tumors belies their WHO 2016 classification at IDH-wild type [27, 28], a distinction generally associated with unfavorable clinical outcome. Accordingly, WHO 2021 integrates defining molecular alterations in the establishment of two additional diagnostic categories: diffuse astrocytoma, MYB or MYBL1-altered, and diffuse low-grade glioma, MAPK pathway-altered. Both entities are IDH1/2 and histone H3-wild type by definition. The former invariably harbors alterations involving the MYB and MYBL1 genes, usually fusions, and has been described in prior work as “isomorphic astrocytoma” or “isomorphic diffuse glioma” [29]. The MYB and MYBL1 fusions characterizing this tumor exhibit an array of partner genes that, interestingly, does not typically include QKI, the most common MYB fusion partner in the related entity of angiocentric glioma (see below). As its name implies, diffuse low-grade glioma, MAPK pathway-altered, is defined by its low-grade infiltrating glial morphology coupled with genomic abnormalities involving MAPK pathway constituents, primarily the BRAF V600E mutation and abnormalities in FGFR1 (tyrosine kinase domain duplication, mutation, or fusion). Additional abnormalities involving FGFR2, NTRK1-3, MAP2K1 and MET, can also rarely be seen [27, 28].
Angiocentric Glioma
This entity was initially established in WHO 2016 [3]and features no significant changes in WHO 2021. Angiocentric gliomas are uncommon tumors composed of monomorphic bipolar cells and characterized by an angiocentric growth pattern featuring ependymal differentiation (Fig. 6a, b). These low-grade neoplasms typically occur in or adjacent to the cerebral cortex and are associated with epilepsy. MYB rearrangements, in particular the MYB-QKI fusion, are almost invariably present. Angiocentric gliomas exhibit indolent clinical behavior corresponding to WHO grade 1 classification.

Angiocentric glioma. These tumors are composed of monomorphic bipolar glial cells (a) and classically exhibit and angiocentric growth pattern reminiscent of ependymoma (b). 200 × magnification
Polymorphous Low-Grade Neuroepithelial Tumor of the Young (PLNTY)
These epileptogenic cerebral tumors occur primarily in children and young adults, although cases in older individuals have been reported, and tend to follow an indolent clinical course. They are characterized histologically by a predominantly diffuse pattern of growth, often featuring oligodendroglioma-like components and calcification, and exhibit extensive expression of CD34 in both tumor cells and ramified neural elements in the associated cerebral cortex [1••, 30]. Astrocytic components are also present in many cases; mitoses are rare to absent. A defining DNA methylation signature delineated PLNTY from other low-grade neuroepithelial neoplasm like ganglioglioma and pilocytic astrocytoma. Additionally, these tumors harbor MAPK pathway-activating alterations, predominantly fusions involving FGFR2 and FGFR3 or BRAF V600E mutations [1••, 30]. Despite their often prominent oligodendroglial morphology, PLNTYs do not harbor IDH1/2 mutation or 1p/19q codeletion.
Conclusions
The integration of defining molecular features into brain tumor diagnosis that began in WHO 2016 only intensifies in WHO 2021. This approach, epitomized by the manifold refinements of diffuse glioma classification discussed above, allows for more granular separation of distinct biological and clinical entities previously grouped together, enabling more effective communication of both diagnostic and prognostic information, and in some instances, particularly for pediatric diffuse gliomas, suggests possible targeted therapies to improve disease management. As this process continues in the ensuing years, additional refinements must be anticipated, and emerging novel entities firmly grounded in standard clinical practice. Accordingly, the importance of ongoing projects like the cIMPACT-NOW series cannot be overstated, along with educational outreach from the neuropathology community. WHO 2021 makes great advances, but it represents only the first step of an iterative process that will repeatedly shift the landscape of neuro-oncology in the foreseeable future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30(4):844–56. https://doi.org/10.1111/bpa.12832. This article details recommendations for the upcoming WHO 2021 Classification of CNS Neoplasms, including refined diagnostic classes and conversion to Arabic numerals for grading.
Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139(3):603–8. https://doi.org/10.1007/s00401-020-02127-9.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D et al. WHO Classification of Tumors of the Central Nervous System. World Health Organization Classification of Tumours. Lyon: International Agency for Research on Cancer; 2016.
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018;20(1):66–77. https://doi.org/10.1093/neuonc/nox132.
• Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019;21(12):1519–28. https://doi.org/10.1093/neuonc/noz124. This article identifies CDKN2A deletion as a robust prognostic marker in IDH-mutant astrocytoma.
• Reis GF, Pekmezci M, Hansen HM, Rice T, Marshall RE, Molinaro AM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol. 2015;74(5):442–52. https://doi.org/10.1097/NEN.0000000000000188. This article identifies CDKN2A deletion as a robust prognostic marker in IDH-mutant astrocytoma.
Banan R, Stichel D, Bleck A, Hong B, Lehmann U, Suwala A, et al. Infratentorial IDH-mutant astrocytoma is a distinct subtype. Acta Neuropathol. 2020;140(4):569–81. https://doi.org/10.1007/s00401-020-02194-y.
•• Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018;136(5):805–10. https://doi.org/10.1007/s00401-018-1913-0. This article establishes molecular inclusion criteria for the diagnosis of glioblastoma, IDH-wild type, WHO grade 4.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77. https://doi.org/10.1016/j.cell.2013.09.034.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31. doi:https://doi.org/10.1038/nature10833.
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–3. https://doi.org/10.1038/ng.1102.
• Louis DN, Giannini C, Capper D, Paulus W, Figarella-Branger D, Lopes MB, et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma. IDH-mutant Acta neuropathologica. 2018;135(4):639–42. https://doi.org/10.1007/s00401-018-1826-y. This article refines criteria for the diagnosis of diffuse midline glioma, with acknowledgement of H3 K27M mutations in other tumor types.
Gessi M, Capper D, Sahm F, Huang K, von Deimling A, Tippelt S, et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol. 2016;132(4):635–7. https://doi.org/10.1007/s00401-016-1608-3.
Hochart A, Escande F, Rocourt N, Grill J, Koubi-Pick V, Beaujot J et al. Long survival in a child with a mutated K27M-H3.3 pilocytic astrocytoma. Annals of clinical and translational neurology. 2015;2(4):439–43. doi:https://doi.org/10.1002/acn3.184.
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–12. https://doi.org/10.1038/ng.2611.
Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015;130(6):815–27. https://doi.org/10.1007/s00401-015-1478-0.
Castel D, Kergrohen T, Tauziede-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3–K27M mutation. Acta Neuropathol. 2020;139(6):1109–13. https://doi.org/10.1007/s00401-020-02142-w.
Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013;23(5):558–64. https://doi.org/10.1111/bpa.12042.
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–37. https://doi.org/10.1016/j.ccr.2012.08.024.
Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016;131(1):137–46. https://doi.org/10.1007/s00401-015-1493-1.
Bressan RB, Southgate B, Ferguson KM, Blin C, Grant V, Alfazema N et al. Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell. 2021;28(5):877–93 e9. doi:https://doi.org/10.1016/j.stem.2021.01.016.
Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF et al. Histone H3.3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell. 2020;183(6):1617–33 e22. doi:https://doi.org/10.1016/j.cell.2020.11.012.
•• Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. 2017;32(4):520–37e5. doi:https://doi.org/10.1016/j.ccell.2017.08.017. This article represents a broad molecular annotation of pediatric high-grade glioma, detailing the spectrum of defining molecular alterations across prospective diagnostic entities.
Guerrini-Rousseau L, Varlet P, Colas C, Andreiuolo F, Bourdeaut F, Dahan K et al. Constitutional mismatch repair deficiency-associated brain tumors: report from the European C4CMMRD consortium. Neurooncol Adv. 2019;1(1):vdz033. doi:https://doi.org/10.1093/noajnl/vdz033.
Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S, et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020;10(7):942–63. https://doi.org/10.1158/2159-8290.CD-19-1030.
Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019;10(1):4343. https://doi.org/10.1038/s41467-019-12187-5.
Ellison DW, Hawkins C, Jones DTW, Onar-Thomas A, Pfister SM, Reifenberger G, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 2019;137(4):683–7. https://doi.org/10.1007/s00401-019-01987-0.
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131(6):833–45. https://doi.org/10.1007/s00401-016-1539-z.
Wefers AK, Stichel D, Schrimpf D, Coras R, Pages M, Tauziede-Espariat A, et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2019. https://doi.org/10.1007/s00401-019-02078-w.
Huse JT, Snuderl M, Jones DT, Brathwaite CD, Altman N, Lavi E, et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. 2017;133(3):417–29. https://doi.org/10.1007/s00401-016-1639-9.
Acknowledgements
We would like to thank Dr. Greg Fuller for providing histopathological images.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuro-Oncology
Rights and permissions
About this article

Cite this article
Perez, A., Huse, J.T. The Evolving Classification of Diffuse Gliomas: World Health Organization Updates for 2021. Curr Neurol Neurosci Rep 21, 67 (2021). https://doi.org/10.1007/s11910-021-01153-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s11910-021-01153-8