
Abstract
Traumatic brain injury (TBI) is a significant cause of mortality and morbidity worldwide. Current treatment of acute TBI includes surgical intervention when needed, followed by supportive critical care such as optimizing cerebral perfusion, preventing pyrexia, and treating raised intracranial pressure. While effective in managing the primary injury to the brain and skull, these treatment modalities do not address the complex secondary cascades that occur at a cellular level following initial injury and greatly affect the ultimate neurologic outcome. These secondary processes involve changes in ionic flux, disruption of cellular function, derangement of blood flow and the blood-brain barrier, and elevated levels of free radicals. Over the past few decades, numerous pharmacologic agents and modalities have been investigated in an attempt to interrupt these secondary processes. No neuroprotective agents currently exist that have been proven to improve neurologic outcome following TBI. However, these trials have contributed significantly to the understanding of the clinical sequelae of TBI and to improvements in the quality of care for TBI. With the experience and insights that have been accrued with the trials to date, we will be able to optimize future trial designs and refine established neurologic endpoints to better identify new therapeutic agents and further improve neurologic outcomes from this often devastating condition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a significant cause of morbidity and mortality worldwide. In the USA alone, over 1.7 million people suffer a TBI every year, with over 50,000 deaths and at least one third of survivors facing long-term disability [1•, 2, 3]. There is a significant physical, emotional, and economic burden on patients and families, as well as on society as many survivors of severe TBI remain disabled for extended periods of time. Research over the past few decades has improved our understanding of the pathophysiology of TBI and has improved outcomes based on management guidelines and treatment paradigms. Advances in trauma systems, imaging and neuro-monitoring techniques have also improved treatment and outcomes [4]. However, despite such progress including enhanced prevention measures, early identification of injury, and prompt resuscitation, mortality remains at about 20–30 % with varying degrees of morbidity in survivors.
There have been numerous studies of traumatic brain injury over the past two decades. These clinical trials have been generally based on laboratory studies that provided the experimental support to advance these pharmacotherapeutic agents into clinical trials [1•, 5, 6]. However, despite success in the laboratory setting and sometimes in early clinical studies, the definitive phase 3 trials have so far been disappointing.
TBI consists of an initial physical injury (primary injury) that results from the impact and often results in skull fractures, cerebral contusions, diffuse axonal injury, and epidural or subdural hematomas. The initial physical damage to the brain tissues is immediate and to a significant extent irreversible. However, following the primary injury, there is a complex cascade of cellular and molecular reactions that occur over a period of days to weeks that result in changes in ionic homeostasis (calcium, potassium, sodium etc.), release of excitatory neurotransmitters and free radicals, and inflammatory and immune reactions, among others [1•, 7•, 8]. These processes offer potential targets for therapeutic intervention. Furthermore, secondary clinical sequelae such as hypotension, hyperthermia, seizures, edema and hypoxia, ischemia, and malignant intracranial hypertension may and often do worsen the ultimate outcome. Diffuse axonal injuries can result from both primary and secondary injuries at the neuronal level [8].
Patients who present with TBI receive a combination of surgical and medical therapies. Mass lesions and depressed fractures are treated surgically, and supportive therapies help maintain cerebral perfusion and oxygenation. Control of intracranial pressure (ICP) and cerebral perfusion pressure as well as systemic oxygenation are key goals of treatment [9]. Other interventions such as early nutritional supplementation, fluid balance, and prevention of complications such as hyperthermia, hyponatremia, seizures, pneumonia, venous thromboembolism, and bleeding all help improve overall morbidity. Since the primary injury cannot be reversed, significant research has been focused on preventing or minimizing secondary injury. A number of therapeutic strategies have been explored, but no neuroprotective agent has been confirmed so far as being effective in the clinical setting.
Physiological background
There are both extracellular and intracellular processes that contribute to secondary injury. In the acute and subacute periods following TBI, there is a significant increase in excitatory neurotransmitter release. Cellular membranes may be damaged by oxidation and ion flux. Concurrently, the immune system’s response to injury also may have a deleterious effect on the injured cells. The combination of cellular metabolic derangement, oxidative stress, and numerous other systemic factors form a complex set of variables that contribute to the secondary injury following TBI.
Disruption of cerebral blood flow autoregulation is common following TBI. Hypotension can therefore be devastating in these patients and maintenance of cerebral blood flow is a key aspect of early TBI treatment [9, 10, 11•]. Blood flow is reduced immediately following TBI due to vasoconstricting prostaglandin release as well as reduced brain oxygen demand due to the injury. However, soon thereafter, cellular acidosis, increases in bradykinin and nitric oxide synthetase, and increases in excitatory neurotransmitters can lead to vasodilatation [12]. Hyperemia and cerebral edema can both result in increased intracranial pressure (ICP) [13].
Ischemia may result from a mismatch between cerebral blood flow and cerebral metabolic rate [14, 15]. Advances in bedside monitors such as intraparenchymal brain tissue oxygen monitors (e.g., Licox) have helped in the detection and treatment of cerebral hypoxia. Mass-occupying lesions such as subdural hematoma or intracerebral hemorrhage may compress cerebral vasculature and cause further ischemia. Surgical decompression will help relieve compression but may also result in reperfusion injury and result in oxidative damage [1]. About 50 % of severe TBI patients may experience vasospasm and are at further risk of ischemia [16].
Inflammatory responses to TBI can be disruptive as well. Pro-inflammatory mediators such as tumor necrosis factor (TNFα) and interleukin-6 (IL-6) are found in high concentrations in injured brain tissue, although their exact roles remain poorly understood [17, 18]. These cytokines promote edema formation and immune activation. Complement cascades similarly act to increase edema, attract immune cells, promote cell lysis, and blood-brain barrier disruption [19]. Blood-brain barrier disruption in turn permits the passage of proteins and other compounds that may alter brain osmolarity and promote edema. The increased permeability of the blood-brain barrier may provide an opportunity for potential neuroprotective agents to penetrate to brain tissues where they are normally excluded and may result in higher brain concentrations. The modulation of the immune system-mediated inflammatory response holds promise in treating neurologic injury.
Oxidation is increased within the brain following TBI. Elevated concentration of reactive species such as superoxide and peroxynitrite can cause DNA damage and lipid peroxidation, which in turn increases inflammation and oxidation in nearby tissues [20, 21]. This promotes apoptosis in the affected cells. The breakdown of the cellular membrane can permit cellular swelling as water moves into the cell. Overexpression of aquaporin may exacerbate cerebral edema as does the dysregulation of ionic homeostasis [22]. Intracellular sodium and calcium concentrations are increased, which leads to increased extracellular potassium concentrations. This is likely due to malfunction of ionic exchange pumps [23].
Calcium can affect the progression of secondary injury and edema following TBI. Increased concentrations of excitatory neurotransmitters such as glutamate can increase the influx of calcium and sodium into cells [17]. High intracellular concentrations of calcium promote mitochondrial cytochrome C and calpain/caspase-mediated apoptosis [24]. As mitochondria degenerate, ATP production drops and cellular activity decreases, ultimately leading to cell death.
Review of selected neuroprotective agents
We will now briefly review the major pathways that have been studied preclinically or have been advanced to clinical testing and if possible will provide observations on the potential explanations for their failure.
Calcium-channel antagonists
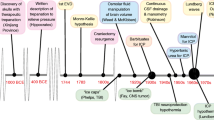
The blocking of neural calcium channels could potentially reduce the deleterious effects of excitatory neurotransmitters and interrupt the apoptosis cascade. Calcium-channel blockers such as nimodipine have been shown to reduce vasospasm in aneurysmal subarachnoid hemorrhage and have been theorized to have a neuroprotective role as well [1•, 5, 25]. Drawbacks to such agents are systemic effects such as hypotension and vasodilatation which could adversely affect patients with TBI. Several TBI trials with calcium-channel blockers such as nimodipine and nicardipine have failed to demonstrate efficacy. The HIT-I trial in 1990 was a randomized study of 350 patients that examined the effect of nimodipine in patients with severe head injury No statistical benefit was found [26]. A larger European study (HIT-II) seemed to show benefit in a subgroup of patients who had traumatic subarachnoid hemorrhage, but a pooled analysis of both HIT trials did not show any benefit of nimodipine in any group [27]. A Cochrane meta-analysis showed some reduction in death and disability after treatment with calcium-channel blockers in patients with traumatic subarachnoid hemorrhage [25]. Nevertheless, treatment with such medications has not seen widespread use in the management of TBI.
Steroids
Steroids have been used to control cerebral edema in numerous neurological conditions such as tumors or inflammatory disorders. There have been nearly 20 clinical trials over the past 40 years investigating steroids for the treatment of TBI, with nearly all of them being underpowered and demonstrating little to no positive effect in improving outcome [28•]. In 1976, Gobiet et al. showed some benefit in administering high-dose dexamethasone to patients with traumatic brain injury [29]. Cooper et al. conducted a prospective double-blinded trial that showed improved 6-month mortality with steroid use in 76 patients with TBI, but showed no change in favorable functional outcome [30]. Additional clinical trials over the years have showed trends toward possible improved outcomes, but were unable to demonstrate clear statistical benefit. Grumme et al. conducted a larger multicenter randomized trial with 396 TBI patients given triamcinolone. A trend toward improved outcomes in the steroid group was seen after injury, yet there was no statistical difference between the two groups after 1 year [31]. Gaab et al. showed no significant improvement in a prospective randomized controlled trial with high-dose dexamethasone administered within 3 h of injury [32].
The CRASH trial was a large simple controlled trial completed in 2005, in which 10,000 TBI patients with a Glasgow coma scale (GCS) ≤14 were randomized to methylprednisolone or placebo within 8 h of injury [33]. Instead of a beneficial effect, the risk of death or severe disability was found to be higher in the steroid group. These data provided the metaphorical “last nail in the coffin” for the use of steroids in the treatment of TBI [34].
NMDA-receptor antagonists
High concentrations of the excitatory neurotransmitter glutamate can destroy neurons. It has been shown that high concentrations of glutamate [100–500 μM] can induce cell death in vitro and that similar extracellular concentrations are present in the rodent brain and spinal cord during ischemia or trauma [5, 6, 7•, 8, 35]. It was therefore hypothesized that glutamate N-methyl-d-aspartate (NMDA) receptor antagonists should improve outcomes after TBI and stroke. Preliminary animal work was done in both ischemic stroke and various models of TBI. Some of the TBI models showed neuroprotective effects only if administered before injury but not following injury [36]. Several clinical trials involving NMDA receptor antagonists such as aptiganel, dextromathorphan, dizocilpine, eliprodil, gavestinel, licostinel and selfotel were initiated, only to be halted prematurely, or failed to demonstrate efficacy in clinical trials of stroke or traumatic brain injury. The results of these same trials were either inadequately reported or never reported at all in the scientific literature (aptiganel, eliprodil, and EAA). Indeed, some trial data suggested that agents in this class may in fact have had a neurotoxic effect [35].
Glutamate agonists
Glutamate receptors are highly expressed on microglia, astrocytes, as well as on neurons. Glutamate agonists have been found to inhibit caspase-dependent apoptosis in many in vitro and in vivo models. They have also been found to attenuate microglial inhibition of NADPH oxidase. Early treatment with glutamate agonists in the laboratory setting showed good neuroprotection after TBI. Fhe mGluR5 agonist, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), is neuroprotective and has anti-apoptotic properties in neuronal cell culture and microglial culture models. Loane et al. have also shown good in vivo neuroprotection results in animal models, and current clinical trials are underway [37].
The NMDA partial agonist d-cycloserine has been shown to be beneficial in animal models as well. Adeleye et al. have shown good results in a mouse study with d-cycloserine administration within 24 h of TBI [38]. The role of glutamate in TBI is interesting in that both glutamate antagonists and agonists have been shown to impart benefit in various models. In a laboratory study in rats, Fei et al. [39] found that both glutamate antagonists and agonists improved neurological severity score, reduced cerebral edema, and decreased neuronal loss following induced TBI. These opposite effects may reflect the unique sensitivity of the nervous system.
Oxygen free-radical scavengers
A multicenter double-blinded randomized trial involving two different doses of pegorgotein vs. placebo following TBI was conducted in the mid-1990s [40]. Four hundred sixty-three patients were enrolled, and there was no statistically significant difference between the two groups in neurologic outcome or mortality at 3 or 6 months after TBI. Post hoc analysis of patients with GCS > 3 showed a trend toward favorable outcomes in patients who were given the lower dose of pegorgotein (10,000 units/kg), though this difference was not significant. The trial itself was underpowered, given the three different treatment arms, but despite this and the potentially favorable outcomes, pegorgotein was ultimately abandoned as a treatment for TBI.
Tirilazad is an aminosteroid with antioxidant and lipid peroxidation inhibition properties and was studied in a large multicenter TBI trial conducted by Marshall et al., enrolling a cohort of 1120 head injury patients of whom 85 % had suffered a severe TBI (GCS 4–8) [41]. There was no difference in favorable outcomes or mortality between treatment and placebo groups. Though there was a mortality benefit in male patients with traumatic subarachnoid hemorrhage, tirilazad was not considered for further investigation and treatment for TBI.
Immune system modulation
Cyclosporin-A is an immunosuppressant drug that impairs T cell-mediated immunity and is used in patients who have received organ transplants. It inhibits calcineurin and cyclophilin-A, as well as inhibits mitochondrial pore formation, which could help inhibit the apoptotic cascade [42]. Several phase 2 trials have demonstrated safety of cyclosporin-A. Hatton et al. conducted a single-center trial administering cyclosporin or placebo to 40 subjects with severe TBI within 8 h of injury [43]. There were no differences in safety measures such as seizure or infection. Patients who received higher doses of cyclosporine seemed to have a greater probability of favorable outcome. A larger randomized controlled trial on 100 patients with GCS < 10 and radiological evidence of DAI showed that cyclosporine had no adverse effects following TBI but did not demonstrate improvement in outcome or mortality [44]. Other studies have demonstrated that there is little effect on lymphocyte count or infection rate after administration in TBI patients [43, 45, 46]. Cyclosporine may yet hold promise for treatment of TBI; a phase 3 trial is currently underway in Canada, and the Copenhagen Head Injury Cyclosporin (CHIC) study is currently recruiting patients as well [47].
Another lymphocyte modulator, fingolimod, inhibits lymphocyte release from lymph nodes and limits lymphocyte circulation. It is FDA approved to treat relapsing multiple sclerosis. Fingolimod helps enhance blood-brain barrier integrity and, in animal models, has been shown to reduce infarction size and promote cell regeneration. A study by Fu et al. in 22 patients with anterior circulation strokes found better neurologic outcomes in patients given fingolimod compared to control [48]. There was a significant reduction of lesion enlargement, microvascular permeability, and better clinical outcomes both during the acute phase and 3-month follow-up. In evaluation of flow cytometry, acute reductions in CD4+ and CD8+ T cells as well as CD19+ B and CD56+ NK cells compared to matched controls which returned to baseline levels in follow-up. In perhaps a more applicable study, Fu et al. have demonstrated in intracerebral hemorrhage patients (n = 23), in an open label design, that similar improvements, assessed by blinded evaluators, were seen in fingolimod-exposed patients, with better neurologic status, smaller hematoma volume, and reduced peri-hematomal volumes [49]. These results are promising and may be applicable to future larger trials for TBI.
Statins
Statins are a widely used class of medications that inhibit HMG-CoA reductase to decrease cholesterol synthesis, lowing serum low-density lipoprotein levels. They seem to have numerous secondary effects, and a well-known safety profile. Statins such as simvastatin, atorvastatin, and rosuvastatin have been successfully used to combat cardiovascular disease and hyperlipidemia. There is evidence of anti-inflammatory properties and immune system activity modulation by reduced leukocyte adhesion [50]. Statins have also been found to improve cerebrovascular function by attenuating endothelial nitric oxide synthetase activity, decreased neuronal apoptosis, and increased production of VEGF and BDNF [51].
There have been numerous in vitro and animal studies exhibiting a neuroprotective benefit of statins in TBI and ischemic stroke [52]. A small phase II study was conducted with 22 patients with moderate TBI where patients received rosuvastatin and were followed for 3 months [53]. There seemed to be cognitive benefits within 2 weeks in the patients who had received the statin. Further studies have shown improvement in disability scores and suggest an anti-inflammatory effect after TBI [54]. However, a larger clinical trial examining the effects of statins in TBI has not yet been performed, though there is currently another phase II trial underway evaluating the effect of atorvastatin administered for 1 week following TBI on cognitive outcomes (NCT01013870).
Progesterone
The neuro-steroid progesterone has been one of the most promising neuroprotective agents for TBI. It was discovered that gender and menstrual cycle seem to confer a favorable outcome benefit after experimental TBI in animals. Progesterone appears to have beneficial effects in the injured brain by limiting edema by reducing lipid peroxidation, aquaporin gene/protein expression, pro-inflammatory cytokine release, and complement activation [55]. These findings led to great interest in testing this hormone in the human setting.
Two phase 2 clinical studies suggested significant efficacy of progesterone in TBI with no significant safety issues. The Progesterone for the Treatment of Traumatic Brain Injury (ProTECT) trial was a NIH-funded phase II trial conducted at a single institution (Emory) in which 100 patients with moderate to severe TBI were randomized to receive progesterone or placebo (3:1 randomization ratio) within 11 h of injury. The safety profile was similar between the two treatment groups. The progesterone-treated group had a significantly lower risk of death within 30 days [56]. A similar phase 2 clinical trial conducted in China enrolled at a single trial site, 159 patients, and those receiving progesterone had a lower mortality rate and a higher incidence of favorable outcomes as measured by the Glasgow outcome score [57]. These two small phase II trials led to the initiation of two large phase 3 trials of progesterone, albeit with slightly different formulations, severity profiles, and time to treatment.
The first of these phase 3 trials, the ProTECT-III trial, was a prospective randomized trial of progesterone in moderate and severe TBI that was conducted with NIH support in the USA. The treatment window was 4 h. The ProTECT-III trial was halted by the Data Safety Monitoring Board (DSMB) after the randomization of 882 patients after a futility analysis demonstrated no benefit [58]. The second phase 3 trial, the SYNAPSE trial, was a multinational prospective randomized controlled trial with 1195 severe TBI patients who were randomized to receive either progesterone or placebo within 8 h of injury. There was no significant difference in safety, outcome, or mortality [59]. These were obviously disappointing outcomes as progesterone appeared to have strong pre-clinical and reasonable but limited data from the phase 2 trials suggesting clinical evidence of efficacy for the treatment of TBI.
Hypothermia
As most neuroprotective agents have proven to be ineffective in large clinical trials, the largest contributions to improved morbidity and mortality following TBI has been attributed to improved resuscitation and critical care. Systemic hypothermia has been investigated as a possible technique to improve outcomes in TBI patients. Mild hypothermia is defined as cooling the body core temperature to approximately 33–34 °C with the intention of decreasing cerebral edema and swelling and to also slow the secondary injury cascade following TBI [60, 61]. In animal models, early administration of hypothermic treatment reduced ICP and cerebral edema [62]. It was anticipated that inducing early hypothermia combined with modern resuscitation and critical care methods could improve outcomes in TBI patients.
In a 2001 multicenter trial, Clifton et al. studied the effects of hypothermia induced in TBI patients within 6 h after a closed head injury. They found that on the background of standard treatment that hypothermia when compared to normothermia resulted in fewer patients with high ICPs in the hypothermic treatment group, although patients in the hypothermic group had longer hospital stays and more medical complications. However, there were no significant differences in a 6-month functional status between the two groups [63]. Post hoc analyses demonstrated a trend toward improved outcomes in patients who presented to the hospital already hypothermic. A second multicenter trial was initiated in which patients had hypothermic therapy initiated within 2.5 h of TBI. The trial was terminated early due to futility [64]. More recently, the Eurotherm3235 trial evaluated the effects of hypothermia in TBI patients with ICPs greater than 20 mmHg. Worse results were reported in the hypothermia group than in the normothermia group [65]. Based on the results from these trials, it is becoming increasingly apparent that although hypothermia appears to have a robust neuroprotective effect in the laboratory, its systemic complications may negate any clinical benefit in patients. Providing localized hypothermia to the brain and avoiding systemic hypothermia have so far proven to be technically challenging.
Causes of trial failures
There has been much speculation as to the causes of trial failure in TBI [26]. An important factor is arguably the proper selection of drugs to take to clinical trial. The implementation of The Stroke Therapy Academic Industry Roundtable (STAIR) proposed working guidelines [66] to improve the utility of preclinical studies has been suggested to form the foundation prior to the initiation of clinical trials in neurological disease and may better inform the translational steps from animal to man. Unfortunately, these principles were unavailable prior to 1999 and thus many of the therapeutics that entered clinical development did not take full advantage of these guidelines. This may have contributed to many of the clinical trial failures described above and has recently been updated [67]. Some of the underlying issues that may have contributed to the ultimate clinical trial failure are listed in Table 1.
There is a clear need to continue to refine our processes and trial designs in order to optimize the opportunity to identify a therapeutic agent(s) that are effective in patients with TBI. In this vein, the NIH has funded a consortium of high-quality TBI basic laboratories that are charged with screening all proposed drugs in multiple animal models. Only if and when all of the laboratories show efficacy in multiple models of TBI will a drug be taken forward for clinical testing.
To further advance the goal of developing treatments for TBI, two consortia are collecting large amounts of pre-specified data on TBI patients in multiple centers in the USA and Europe [68–70]. One idea behind this “big data” approach is to design a better classification of TBI than the simplistic “mild, moderate, and severe” classification. The other is to identify better correlates of outcome that may allow us to find alternative trial designs that are more sensitive in specific subgroups of patients.
Finally, it should be noted that the overall number of trials conducted in TBI pale in comparison to the number of trials conducted in various cancers. For a condition with such great public health significance, TBI remains a grossly understudied disease.
Conclusions
The outcomes from severe traumatic brain injury have improved substantially over the past few decades—from a mortality of around 50 % in the 1970s to about half of that in current series. Furthermore, this enhanced survival has modified the outcome of the “vegetative” or “severely disabled” patients, who have gone on to make a “good” or “moderately disabled” recovery. However, this remarkable improvement cannot be attributed to any single drug. Several pharmacological approaches have been tested with increasing sophistication of trial design. However, for the most part, these trials have failed to show a beneficial effect. In this paper, we have briefly reviewed the different drug classes that have been tested and the rationale for doing so. Trials to date have mostly depended on 6-month outcomes as the primary endpoint. Phase 3 trials in this patient population typically call for approximately 1000 patients, take 3–5 years to complete, and cost multiple millions of dollars. We have not yet identified any biomarkers that could serve as surrogate indicators of drug efficacy. If identified, such surrogates could potentially simplify the early screening of drugs in development leading to large trials for only those drugs that have demonstrated a clear pharmacological signal in the clinical setting.
References
Papers of particular interest, published recently, have been highlited as: • Of importance
McConeghy KW, Hatton J, Hughes L, Cook AM. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs. 2012;26(7):613–36. An excellent review of neuroprotection pharmacology.
Thurman D, Guerrero J. Trends in hospitalization associated with traumatic brain injury. JAMA. 1999;282(10):954–7.
Pearson WS, Ovalle Jr F, Faul M, Sasser SM. A review of traumatic brain injury trauma center visits meeting physiologic criteria from The American College of Surgeons Committee on Trauma/Centers for Disease Control and Prevention Field Triage Guidelines. Prehosp Emerg Care. 2012;16(3):323–8.
Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, et al. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill Summ. 2011;60(5):1–32.
Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7(1):115–26.
Vink R, Van Den Heuvel C. Recent advances in the development of multifactorial therapies for the treatment of traumatic brain injury. Expert Opin Investig Drugs. 2004;13(10):1263–74.
Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci. 2014;15(1):1216–36.
A review of the of neuroprotection strategies and how to improve the translation of animal models 8, Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Curr Pharm Des. 2001;7(15):1475–503.
Myburgh J, Cooper DJ, Finfer S, Bellomo R, Norton R, Bishop N, et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357(9):874–84.
Manley G, Knudson MM, Morabito D, Damron S, Erickson V, Pitts L. Hypotension, hypoxia, and head injury: frequency, duration, and consequences. Arch Surg. 2001;136(10):1118–23.
Czosnyka M, Smielewski P, Piechnik S, Steiner LA, Pickard JD. Cerebral autoregulation following head injury. J Neurosurg. 2001;95(5):756–63. A concise review of the physiological basis of the brain’s response to injury.
Steiner LA, Coles JP, Johnston AJ, Chatfield DA, Smielewski P, Fryer TD, et al. Assessment of cerebrovascular autoregulation in head-injured patients: a validation study. Stroke. 2003;34(10):2404–9.
Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol. 2004;14(2):195–201.
Obrist WD, Langfitt TW, Jaggi JL, Cruz J, Gennarelli TA. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg. 1984;61(2):241–53.
Overgaard J, Tweed WA. Cerebral circulation after head injury. 1. Cerebral blood flow and its regulation after closed head injury with emphasis on clinical correlations. J Neurosurg. 1974;41(5):531–41.
Oertel M, Boscardin WJ, Obrist WD, Glenn TC, McArthur DL, Gravori T, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103(5):812–24.
Enriquez P, Bullock R. Molecular and cellular mechanisms in the pathophysiology of severe head injury. Curr Pharm Des. 2004;10(18):2131–43.
Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab. 2014;34(5):845–51.
Ott L, McClain CJ, Gillespie M, Young B. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11(5):447–72.
Hall ED, Andrus PK, Yonkers PA. Brain hydroxyl radical generation in acute experimental head injury. J Neurochem. 1993;60(2):588–94.
Shohami E, Beit-Yannai E, Horowitz M, Kohen R. Oxidative stress in closed-head injury: brain antioxidant capacity as an indicator of functional outcome. J Cereb Blood Flow Metab. 1997;17(10):1007–19.
Venero JL, Machado A, Cano J. Importance of aquaporins in the physiopathology of brain edema. Curr Pharm Des. 2004;10(18):2153–61.
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22(5):E1.
Young W. Role of calcium in central nervous system injuries. J Neurotrauma. 1992;9 Suppl 1:S9–25.
Langham J, Goldfrad C, Teasdale G, Shaw D, Rowan K. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst Rev. 2003;4:CD000565.
Teasdale G, Bailey I, Bell A, Gray J, Gullan R, Heiskanan O, et al. A randomized trial of nimodipine in severe head injury: HIT I. British/Finnish Co-operative Head Injury Trial Group. J Neurotrauma. 1992;9 Suppl 2:S545–50.
Murray GD, Teasdale GM, Schmitz H. Nimodipine in traumatic subarachnoid haemorrhage: a re-analysis of the HIT I and HIT II trials. Acta Neurochir (Wien). 1996;138(10):1163–7.
Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19(5):503–57. A focused review of clinical trials that provides insights on trial design and endpoints.
Gobiet W. The influence of various doses of dexamethasone on intracranial pressue in patients with severe head injury. Dynamics of brain edema. Montreal: Springer; 1976.
Cooper PR, Moody S, Clark WK, Kirkpatrick J, Maravilla K, Gould AL, et al. Dexamethasone and severe head injury. A prospective double-blind study. J Neurosurg. 1979;51(3):307–16.
Grumme T, Baethmann A, Kolodziejczyk D, Krimmer J, Fischer M, von Eisenhart RB, et al. Treatment of patients with severe head injury by triamcinolone: a prospective, controlled multicenter clinical trial of 396 cases. Res Exp Med (Berl). 1995;195(4):217–29.
Gaab MR, Trost HA, Alcantara A, Karimi-Nejad A, Moskopp D, Schultheiss R, et al. “Ultrahigh” dexamethasone in acute brain injury. Results from a prospective randomized double-blind multicenter trial (GUDHIS). German Ultrahigh Dexamethasone Head Injury Study Group. Zentralbl Neurochir. 1994;55(3):135–43.
Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury—outcomes at 6 months. Lancet. 2005;365(9475):1957–9.
Ghajar J, Hesdorffer DC. Steroids CRASH out of head-injury treatment. Lancet Neurol. 2004;3(12):708.
Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1(6):383–6.
McIntosh TK, Vink R, Soares H, Hayes R, Simon R. Effects of the N-methyl-D-aspartate receptor blocker MK-801 on neurologic function after experimental brain injury. J Neurotrauma. 1989;6(4):247–59.
Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604.
Adeleye A, Shohami E, Nachman D, Alexandrovich A, Trembovler V, Yaka R, et al. D-cycloserine improves functional outcome after traumatic brain injury with wide therapeutic window. Eur J Pharmacol. 2010;629(1-3):25–30.
Fei Z, Zhang X, Bai HM, Jiang XF, Wang XL. Metabotropic glutamate receptor antagonists and agonists: potential neuroprotectors in diffuse brain injury. J Clin Neurosci. 2006;13(10):1023–7.
Young B, Runge JW, Waxman KS, Harrington T, Wilberger J, Muizelaar JP, et al. Effects of pegorgotein on neurologic outcome of patients with severe head injury. A multicenter, randomized controlled trial. JAMA. 1996;276(7):538–43.
Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89(4):519–25.
Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79(1-2):231–9.
Hatton J, Rosbolt B, Empey P, Kryscio R, Young B. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg. 2008;109(4):699–707.
Aminmansour B, Fard SA, Habibabadi MR, Moein P, Norouzi R, Naderan M. The efficacy of cyclosporine-A on diffuse axonal injury after traumatic brain injury. Adv Biomed Res. 2014;3:35.
Cook AM, Whitlow J, Hatton J, Young B. Cyclosporine A for neuroprotection: establishing dosing guidelines for safe and effective use. Expert Opin Drug Saf. 2009;8(4):411–9.
Mazzeo AT, Brophy GM, Gilman CB, Alves OL, Robles JR, Hayes RL, et al. Safety and tolerability of cyclosporin A in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26(12):2195–206.
Aertker BM, Bedi S, Cox CS, Jr. Strategies for CNS repair following TBI. Exp Neurol. 2016;275(3):411–26.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315–20.
Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. 2014;71(9):1092–101.
Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci. 2002;23(10):482–6.
Wible EF, Laskowitz DT. Statins in traumatic brain injury. Neurotherapeutics. 2010;7(1):62–73.
Weant KA, Cook AM. Potential roles for statins in critically ill patients. Pharmacotherapy. 2007;27(9):1279–96.
Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, Gordillo-Moscoso A, Martinez-Perez P, Madeville P, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J Neurotrauma. 2008;25(8):1011–7.
Sanchez-Aguilar M, Tapia-Perez JH, Sanchez-Rodriguez JJ, Vinas-Rios JM, Martinez-Perez P, de la Cruz-Mendoza E, et al. Effect of rosuvastatin on cytokines after traumatic head injury. J Neurosurg. 2013;118(3):669–75.
Stein DG. Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev. 2008;57(2):386–97.
Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49(4):391–402. e1-2.
Xiao G, Wei J, Yan W, Wang W, Lu Z. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12(2):R61.
Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371(26):2457–66.
Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, et al. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371(26):2467–76.
Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20(7):904–10.
Jiang JY, Lyeth BG, Kapasi MZ, Jenkins LW, Povlishock JT. Moderate hypothermia reduces blood-brain barrier disruption following traumatic brain injury in the rat. Acta Neuropathol. 1992;84(5):495–500.
Markgraf CG, Clifton GL, Moody MR. Treatment window for hypothermia in brain injury. J Neurosurg. 2001;95(6):979–83.
Clifton GL, Miller ER, Choi SC, Levin HS, McCauley S, Smith Jr KR, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344(8):556–63.
Clifton GL, Valadka A, Zygun D, Coffey CS, Drever P, Fourwinds S, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 2011;10(2):131–9.
Andrews PJ, Sinclair HL, Rodriguez A, Harris BA, Battison CG, Rhodes JK, et al. Hypothermia for intracranial hypertension after traumatic brain injury. N Engl J Med. 2015;373(25):2403–12.
Roundtable STAIR. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30(12):2752–8.
Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–50.
Lingsma HF, Yue JK, Maas AI, Steyerberg EW, Manley GT, Cooper SR, et al. Outcome prediction after mild and complicated mild traumatic brain injury: external validation of existing models and identification of new predictors using the TRACK-TBI pilot study. J Neurotrauma. 2015;32(2):83–94.
Maas AI, Menon DK, Steyerberg EW, Citerio G, Lecky F, Manley GT, et al. Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): a prospective longitudinal observational study. Neurosurgery. 2015;76(1):67–80.
Manley GT, Maas AI. Traumatic brain injury: an international knowledge-based approach. JAMA. 2013;310(5):473–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Shamik Chakraborty declares no conflict of interest.
Brett Skolnick has received an honorarium for serving on the steering committee of BHR Progesterone Trial and a consultancy fee from Pfizer for work on Pfizer’s Factor Xa activities in ICH.
Raj K. Narayan received an honorarium for serving on the steering committee of BHR Progesterone Trial.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neurotrauma
Rights and permissions
About this article

Cite this article
Chakraborty, S., Skolnick, B. & Narayan, R.K. Neuroprotection Trials in Traumatic Brain Injury. Curr Neurol Neurosci Rep 16, 29 (2016). https://doi.org/10.1007/s11910-016-0625-x
Published:
DOI: https://doi.org/10.1007/s11910-016-0625-x