
Abstract
Purpose of Review
Excess sodium from dietary salt (NaCl) is linked to elevations in blood pressure (BP). However, salt sensitivity of BP varies widely between individuals and there are data suggesting that salt adversely affects target organs, irrespective of BP.
Recent Findings
High dietary salt has been shown to adversely affect the vasculature, heart, kidneys, skin, brain, and bone. Common mediators of the target organ dysfunction include heightened inflammation and oxidative stress. These physiological alterations may contribute to disease development over time. Despite the adverse effects of salt on BP and several organ systems, there is controversy surrounding lower salt intakes and cardiovascular outcomes.
Summary
Our goal here is to review the physiology contributing to BP-independent effects of salt and address the controversy around lower salt intakes and cardiovascular outcomes. We will also address the importance of background diet in modulating the effects of dietary salt.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Americans’ predilection for dietary salt has resulted in mean daily sodium (Na+) intake of ~ 3500 mg/day despite multiple organizations recommending ≤ 2300 mg/day [1•, 2]. Recommendations to reduce dietary Na+ are primarily based on studies demonstrating a positive association between Na+ intake and systolic blood pressure (BP) [3, 4]. Clinical trials have demonstrated BP-lowering effects down to a Na+ intake of 1500 mg/day [5]. Recent meta-analyses and systematic reviews of randomized clinical trials (RCTs) have found significant reductions in systolic BP with dietary Na+ restriction, particularly in individuals with hypertension [6,7,8]. In addition, pre-clinical and clinical studies have demonstrated that high Na+ adversely affects multiple target organs independent of BP. Thus, guidelines to avoid high dietary Na+ are supported by a wide range of physiological and clinical studies. However, there is some controversy regarding population-wide efforts to reduce Na+ intake. While there is agreement that high Na+ intake (> 5000 mg/day) is harmful, several cohort studies have demonstrated a paradoxical increase in cardiovascular (CV) events with lower Na+ intake [9•, 10,11,12]. This review article will (1) highlight recent data suggesting that high dietary Na+ can cause target organ damage independent of BP (summarized in Fig. 1); (2) review the epidemiological outcome studies that support Na+ reduction efforts; (3) review the cohort studies that suggest Na+ reduction efforts may have potential negative effects on CV outcomes; and (4) highlight eating patterns that may modulate the relation between salt and CV health.

There is agreement high salt diets contribute to high blood pressure and end organ damage. One of the reasons that consumers have had such a hard time reducing sodium intake is that a majority of the sodium consumed in the diet (> 70%) comes from processed food followed by naturally occurring sodium, and finally what is added in the home. To the right, the various end organ damage associated with high dietary sodium is depicted. The source of the information on sources of dietary sodium is Harnack et al., 2017. [13]
Salt and Target Organ Effects: BP-Independent Effects of Salt
Arteries
Rodent studies demonstrate impaired endothelial function during Na+ loading, without alterations in BP [14,15,16]. Our lab and others have found that high dietary salt adversely affects both large [17,18,19,20,21,22,23] and small [24, 25] artery function in humans. Both salt-resistant and salt-sensitive participants demonstrate impaired endothelial function following high-salt diets, [17] and females seem to be modestly protected compared with males [18, 23]. We recently demonstrated that a high-salt diet increased arterial stiffness in healthy middle-aged adults but the relation was mediated by changes in mean BP [21]. The high-salt diet also elicited greater forward and reflected wave amplitudes [21]. A recent meta-analysis of human trials also suggest that Na+ reduction reduces arterial stiffness [26]. While most of the studies discussed herein were short-term (5-7 days) controlled feeding studies, there are acute feeding studies in humans demonstrating a single high-salt meal impairs endothelial function and increases arterial stiffness [27, 28].
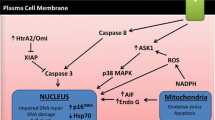
Multiple rodent [14,15,16, 29] and human studies [24, 25] demonstrate that the negative effects of high Na+ on the vasculature are mediated by reactive oxygen species (predominantly superoxide, O2−). High dietary salt results in an increase in O2− which decreases nitric oxide (NO) bioavailability by scavenging NO to form the more stable peroxynitrate radical (ONOO−), or by O2−/ONOO− uncoupling NO synthase [24, 30, 31]. Thus, high dietary Na+ impairs endothelial function through reduced NO bioavailability [32, 33]. In addition to increasing oxidative stress, high Na+ may decrease anti-oxidant defense mechanisms. Specifically, rodent models have demonstrated high Na+ reduces expression of copper/zinc-dependent superoxide dismutase (SOD), which catalyzes the reduction of O2− to hydrogen peroxide [16••, 34]. This can be prevented by administering sub-pressor doses of angiotensin II [34] (which is suppressed during high Na+ diets) and regular exercise [16••]. There are also cellular studies demonstrating hypernatremia results in degradation of the endothelial glycocalyx, which may also contribute to impaired endothelial responsiveness to shear stress [35].
Dietary salt also increases circulating endothelin-1 [27] and arginine vasopressin [36,37,38], two potent vasoconstrictors. A pro-constrictive vasculature is thought to contribute to augmented sympathetic vascular transduction (i.e., vasoconstrictor responses to bursts of sympathetic nerve activity) [39], and we recently demonstrated dietary Na+ restriction (down to 1000 mg/day) reduced sympathetic vascular transduction [40••]. Taken together, several human studies have demonstrated that dietary salt contributes to vascular dysfunction and pre-clinical studies have identified several potential pathways through which this dysfunction may occur.
Kidneys
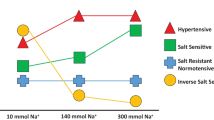
Salt-sensitive hypertension is associated with impaired fluid excretion which contributes to transient increases in cardiac output [41•]. With high salt intake, total peripheral vascular resistance may initially decrease due to reflex/hormonal mechanisms that cause vasodilation (e.g., the baroreflex or suppression of angiotensin II) in response to salt-induced volume loading but normally returns to basal values within several days [41•]. However, renal dysfunction, characterized by impaired pressure natriuresis, has been demonstrated in experimental models of salt-sensitive BP and human salt-sensitive hypertension [41•]. Normally, high levels of dietary Na+ suppress the renin-angiotensin-aldosterone system (RAAS) [17, 18, 42••], contributing to reduced Na+ reabsorption. The epithelial Na+ channel (ENaC), an effector of the RAAS expressed in the distal nephron of the kidney, plays a critical role in regulation of Na+ reabsorption. ENaC has been shown to contribute to increases in serum Na+ and/or plasma volume expansion in the setting of salt-sensitive hypertension [43]. Generally, Na+-driven reabsorption is diminished in response to salt loads; however, in salt-sensitive rodent models, ENaC protein abundance and activity are paradoxically increased, potentially as a result of high-salt-induced renal damage [44, 45]. As discussed in our prior review, salt sensitivity of BP increases with age [46] and aging-related changes in renal Na+ handling may contribute to this increased salt sensitivity [47].
Heart
Salt-sensitive rodent models demonstrate heart failure subsequent to pathological left ventricle (LV) remodeling and the development of hypertension with high Na+ [48]. In humans, LV mass is also associated with hypertension [49, 50]. Independent of BP, high dietary Na+ may increase LV wall thickness and mass [51]. In a longitudinal study of patients who underwent prospective treatment of essential hypertension over three years, LV mass progressively increased across Na+ tertiles only in patients with high plasma aldosterone concentrations. This relation demonstrates a potential interaction between high Na+ consumption and lack of appropriate aldosterone suppression on LV mass [52]. Recent data demonstrate that individuals with masked hypertension have higher Na+ excretion and LV mass [49]. Separately, LV mass was independently associated with Na+ excretion [49]. Another recent study that entailed a prospective analysis of diastolic function and LV mass index in participants with elevated BP found that the dietary Na+ to potassium (K+) ratio was associated with higher LV mass [53]. Estimated Na+ consumption was also positively associated with atrial filling fraction [53]. However, Na+ and K+ intakes were ascertained using a block food frequency questionnaire whereas the other original studies discussed in this section used 24-h urinary Na+ excretion (24hU-Na+); thus, the findings from this final study must be interpreted with caution [54].
Skin and Inflammation
Two recent excellent reviews have highlighted several studies demonstrating Na+ loading results in non-osmotic Na+ accumulation in skin without commensurate increases in skin water content [55••, 56••]. Immune cells including macrophages function as local on-site sensors of interstitial electrolyte concentration and activate tonicity-responsive enhancer binding protein, which increases the expression of vascular endothelial growth factor C (VEGF-C) gene via autocrine signaling [55••, 56••]. VEGF-C facilitates lymphangiogenesis enabling drainage of water and electrolyte from the skin into the systemic circulation for eventual removal via the kidneys [56••]. In rodents, macrophage or VEGF-C antagonism or genetic deletion results in augmented interstitial hypertonic volume retention and elevated BP [57, 58]. Another recent study demonstrated that high salt increased whole body Na+ without increases in body water [59]. Taken together, these findings suggest that non-osmotic Na+ deposition plays a functional role in whole body Na+ homeostasis and BP regulation. However, it has also been shown that T cells exposed to local high-Na+ tissue conditions polarize into highly pro-inflammatory TH17 phenotype cells that produce inflammatory cytokines and worsen experimental autoimmune disease and may contribute to hypertension [60, 61].
In agreement, recent human studies have demonstrated skin Na+ is a marker of aging and hypertension [62]. Excess skin Na+ deposition has also been observed in patients with type 2 diabetes [63] and hyperlipidemia [64], and is associated with cardiac hypertrophy in chronic kidney disease [65]. Further, a recent study randomized healthy participants to consume low- and high-salt diets in a crossover design [66••]. Skin Na+:K+ increased on the high-salt diet in male, but not female participants. Female participants experienced an increase in BP on the high salt, but this may have been confounded by their lower basal BP. Further, in male participants, skin Na+:K+ correlated with BP. There is some thought that increased skin Na+ occurs after sufficient vascular damage resulting in a “leak” into the surrounding tissue [55••]. Using this line of reasoning, the finding that only males experienced an increase in skin Na+ with high salt is supported by prior studies demonstrating that high Na+ damages the endothelial glycocalyx [35] and females are relatively protected against endothelial dysfunction following salt loading compared with males [18, 23]. Nonetheless, more data are needed to elucidate the role of non-osmotic Na+ deposition in humans, and further the influence of dietary Na+ on non-osmotic Na+ deposition. For example, future studies are needed to determine if there are aging and racial differences in skin Na+ with dietary salt manipulation.
Brain Blood Flow and Sympathetic Outflow
In addition to the deleterious effects of dietary salt on peripheral arteries, there is evidence that high Na+ may adversely affect the cerebrovasculature. Classic data from INTERSALT [67] and a prospective Japanese cohort study [68] suggest that high dietary Na+ is associated with increased mortality from strokes. Recent preclinical data from rodents suggest a high-salt diet impairs cerebrovasculature function via excess inflammation and oxidative stress [69, 70]. These findings are consistent with prior studies demonstrating high Na+ reduces expression of SOD and elicits vascular dysfunction in cerebral arteries [34]. Cerebral autoregulation is the ability of the brain to maintain perfusion despite changes in systemic BP, and recent rodent data demonstrate short-term (3 days) and chronic (4 weeks) high salt impairs cerebral autoregulation [71]. Taken together, these cross-sectional human and experimental rodent data are important as impaired cerebrovascular dilatory responses to stimuli in humans are associated with Alzheimer’s disease and stroke incidence [72]. Future RCTs are needed to determine the influence of dietary salt manipulation on cerebrovascular function in humans.
Regarding the effects of salt on sympathetic outflow, salt is thought to sensitize central sympathetic circuits, and excessive sympathetic outflow contributes to the development of CV disease [73]. We recently demonstrated that one week of high salt alters cardiovagal baroreflex sensitivity in healthy adults [74]. Elevations in plasma and cerebrospinal fluid (CSF) Na+ enhance sympathetic nerve activity (SNA) via the rostral ventrolateral medulla (RVLM) leading to increases in BP [75]. Rodent studies demonstrate that blocking SNA attenuates the BP elevations induced by high Na+ in the CSF [75]. One prior human study found that CSF Na+ was elevated following a high-salt diet in salt-sensitive and salt-resistant humans [76]. However, in the group classified salt-resistant, there was a change in mean arterial BP of ~ 5 mmHg (Δ low- to high-salt diet), which many would consider to be salt-sensitive BP [77••]. Thus, more human work is needed in this area. Nonetheless, potential “sensing” mechanisms for Na+ existing in the brain have been elucidated using rodent models [75, 78,79,80]. Central Na+ sensing occurs in the circumventricular organs including the organum vasculosum of the lamina terminalis (OVLT) and subfornical organ, which lack an intact blood-brain barrier [81]. The OVLT has neural connections to the paraventricular nucleus of the hypothalamus, which plays an essential role in regulating SNA outflow via neuronal projections to the RVLM [82, 83]. A newly published study adds additional mechanistic insight, suggesting Nax-positive glial cells in OVLT are activated by high Na+ concentrations, leading to enhanced hydrogen and lactate through a monocarboxylate transporter to activate ASIC1a-positive OVLT neurons [84••].
Bone
While not typically thought of when considering CV health, bone is a target organ that may also be influenced by dietary Na+ intake. There are data indicating high salt intake increases urinary calcium excretion, which may increase risk for osteoporosis [85]. In a cross-sectional study of postmenopausal Korean women, higher urinary Na+:creatinine was positively associated with osteoporosis and negatively correlated with bone mineral density (BMD) [85]. In another cross-sectional Korean study [86], urinary Na+ excretion was negatively associated with bone mineral content (BMC) and BMD in female, but not male participants [86]. In a cross-sectional study of Chinese adults, urinary Na+:K+ was inversely associated with BMD in female, but not male participants [87]. A recent meta-analysis demonstrated that higher Na+ significantly increased the risk of osteoporosis; however, there was a high degree of heterogeneity among studies [88]. Lastly, a prospective observational cohort study of postmenopausal women in the US Women’s Health Initiative demonstrated that there was no association of dietary Na+ intake with changes in BMD at any skeletal site [89]. Collectively, these studies suggest that high dietary salt intake may be associated with impaired BMC, BMD, and risk of osteoporosis; however, there appear to be ethnicity and sex differences. Additionally, controlling for physical activity is an important consideration for studies investigating BMD and BMC. As such, additional RCTs and prospective cohort studies using reliable estimations of Na+ consumption (i.e., controlled feeding and multiple 24hU-Na+ collections) are needed to elucidate the role of dietary salt on bone health.
Dietary Salt and Epidemiology
Controversy Around Dietary Salt and Cardiovascular Outcomes
Some studies have attributed excess dietary salt intake to over 1.5 million CV deaths per year globally due to the fact that dietary salt is linked to increased BP [90]. There is also a large body of data on the relation between BP (a leading risk factor for CV disease) and dietary salt from observational studies and RCTs [91]. Some studies support a linear relation between dietary Na+ intake and CV events. For example, in Trials of Hypertension Prevention (TOHP) phases 1 and 2, Na+ intake was assessed using multiple 24hU-Na+ samples in prehypertensive individuals and participants were followed for an extended period of time (15-year follow-up for TOHP 1 and 10-year follow-up for TOHP 2). There was a 17% increase in CV events for every 1000 mg/day increase in Na+ and no evidence of a J-shaped relationship [5]. A more recent analysis of post-surveillance TOHP also confirmed these findings [92].
Mills et al. [93] investigated the association between urinary 24hU-Na+ excretion (average of three collections) and clinical CV events in patients with chronic kidney disease from the Chronic Renal Insufficiency Cohort (CRIC) Study. The cumulative incidence of CV events in the highest quartile of Na+ excretion was greater compared with the lowest quartile, and the data indicated a significant linear association between Na+ excretion and CV events. However, while both RCT and observational data provide consistent evidence supporting a reduction in dietary sodium intake to below 5000 mg/day, some cohort studies have identified the “optimal” range of Na+ intake for minimizing risk of CV events and mortality to be in the range of ~ 3000–5000 mg/day [10, 11, 93,94,95,96,97,98]. These data are inconsistent with the recommendations put forth by Dietary Guidelines for Americans and the American Heart Association (i.e., < 2300 mg/day) and have been criticized for possible errors, which are detailed below.
As discussed in our prior review [46] and by Cobbs et al. [54] in their 2014 AHA Scientific Advisory on Methodological Issues in Cohort Studies related to dietary Na+ intake and CV disease, errors in study design and statistical power have contributed to some of the controversy. These errors include systematic errors in Na+ assessment (e.g., estimating dietary Na+ intake through food frequency questionnaires, 24-h recall, spot or overnight urine collection). In fact, recent data suggest even 24hU-Na+ may not be satisfactory and that multiple collections are needed to reliably quantify dietary Na+ intake [99]. Additional errors include reverse causality related to recruiting sick patients who may consume low Na+ as part of a therapeutic strategy or reduced overall food consumption or for not excluding sick participants from general population studies.
Studies that have identified the “optimal” range of sodium intake for minimal risk of CV events and mortality to be in the range of ~ 3000–5000 mg/day include several publications from the Prospective Urban Rural Epidemiology (PURE) study, a large epidemiological cohort study spanning 18 countries [9•, 10, 97]. The primary criticism of many of these publications is that participants’ Na+ excretion was estimated from a single fasting morning urine specimen which is problematic because this method overestimates Na+ excretion at lower Na+ levels and underestimates Na+ at higher Na+ levels [100, 101]. Additional critiques include the use of repeated analyses from PURE and the fact that they were conducted by the same research group [102, 103]. However, an additional meta-analysis on dietary Na+ intake and the incidence of CV disease and all-cause mortality also found a U-shaped relation between Na+ intakes and increased mortality [104]. Of note, this analysis included several population samples including smokers, obese individuals and patients with CV risk factors and diabetes and was thus vulnerable to reverse causality. Additionally, dietary Na+ intake was estimated using several assessments including 24-h and spot urine collections, dietary recalls, and food frequency questionnaires likely introducing measurement error.
Some studies that have used 24hU-Na+ have also found an inverse relation between Na+ intake and adverse outcomes (e.g., CV events and mortality); however, these studies were focused on type 2 [95] and type 1 [94] diabetic individuals. As such, these studies include high potential for reverse causality and inadequate covariate adjustment [54]. Taken together with the findings of Mills et al. [93] (who found a beneficial effect of Na+ restriction in CRIC), these studies suggest RCTs are warranted in diabetic patients and those with renal insufficiency to formally test the potential benefit or harm of Na+ restriction in these populations.
In another study using 24hU-Na+ in a healthier study sample, Stolarz-Skrzypek et al. [11] performed a prospective study involving participants without CV disease. Mortality decreased across increasing tertiles of 24hU-Na+. Among the non-hypertensive participants, the risk of hypertension did not increase across increasing tertiles of 24hU-Na+ and there was no relation with overall mortality. Lastly, in a subset of participants who provided multiple 24hU-Na+ samples, a 100-mmol (2300 mg/day) increase in Na+ excretion was associated with an increase in systolic but not diastolic BP. However, this study has been critiqued for inadequate power and follow-up, in addition to a random error in Na+ assessment [54]. To summarize, studies on dietary Na+ and CV outcomes present divergent findings.
Challenges with Dietary Salt Research
Due to the controversy regarding Na+ intake and outcomes, experts in the field of salt and CV health recently called for large-scale dietary salt feeding trials [91, 105••], with some even suggesting the use of a controlled population (i.e., prisoners) [105••], but this is highly controversial. The reason for these proposed studies is that, ideally, investigations on the effects of dietary salt on CV outcomes would largely come from RCTs to reduce potential bias. However, RCTs use interventions that are not feasible for large cohorts given significant practical challenges, including long-term compliance on an assigned dietary salt intake regimen (i.e., several years) and long-term follow-up (post-intervention surveillance) [91, 105••]. Additionally, behavioral intervention trials focused on BP reduction demonstrate that, in free-living people, even modest reductions in dietary salt are difficult to maintain [106]. This difficulty is likely related to the Na+-rich food environment that is common in post-industrial societies.
Dietary Salt Manipulation and the Importance of Lifestyle
Background Diet
Fewer than 10% of Americans adhere to the Dietary Guidelines for Americans dietary Na+ recommendations [1•, 2]. In fact, recent data suggest many Americans consume more than the dietary Na+ recommendation in a single meal when eating at restaurants [107•, 108•]. However, other dietary factors may modulate the relation between Na+ and CV health. For example, a recent review highlighted a large body of literature emphasizing the importance of Cl− in NaCl as having a specific role in salt sensitivity of BP along with Na+ [109•]. This review notes the concept of Cl-mediating physiological responses to Na+ salt dating back to the early twentieth century and highlights several papers demonstrating that other anions for Na+ salt (e.g., citrate, bicarbonate, and phosphate) do not result in elevated BP when equimolar concentration of Na+ are given compared with NaCl in humans [110,111,112,113].
Another consideration with dietary salt intake is its association with overall diet quality. Over 70% of Americans’ dietary Na+ comes from processed food [13] (see Fig. 1). Several epidemiological studies have demonstrated an association between dietary salt intake and obesity, a strong independent risk factor for CV disease, and a common link is thought to be processed food [114]. Ultra-processed foods and dietary salt are both associated with passive overconsumption of calories and poor diet quality [115,116,117]. One recently proposed mechanism from a controlled human feeding study is that high dietary salt was associated with increased fasting ghrelin levels, which could promote increased caloric intake [118]. Another plausible explanation is that high dietary Na+ results in increased muscle catabolism to produce urea as a counter-regulatory mechanism to conserve body water in the context of high-salt diets [59]. This tissue catabolism and subsequent increase in glucocorticoid release may increase appetite [59]. However, another recent study found that 24hU-Na+ is associated with increased overweight/obesity and central adiposity even after adjustment for energy intake and sugar-sweetened beverage consumption [119]. Future longitudinal studies and mechanistic studies are needed to further interrogate the association between high dietary salt consumption and overweight/obesity status.
The importance of background diet in modulating the effects of Na+ also comes from several studies providing high salt in the context of an otherwise healthy diet. A high-salt Dietary Approaches to Stop Hypertension (DASH) diet (i.e., high in fruit, vegetables, low-fat dairy) results in lower BP than a high-salt standard diet [4, 120]. In our previous review, we focused on ethnic disparities in salt sensitivity of BP [46]; we recently demonstrated black individuals are less able to buffer Na+ loads and have greater BP for any given serum Na+ [42••]. There is also some thought that differences in background diet may explain racial differences in salt sensitivity. A recent meta-analysis that included 185 studies suggests salt reduction results in a greater BP reduction in normotensive black individuals compared with normotensive white individuals [11]. In support of background diet mediating these racial differences in BP sensitivity, current Na+ intake appears to be similar across ethnic subgroups [121]; however, large datasets indicate ethnic disparities in diet such that minority groups consume lower amounts of fruit and vegetables, dairy products, and fiber (e.g., whole grains) [122,123,124,125,126,127] while consuming more micronutrient-poor energy-dense foods, meat, and fried food [123]. Also supporting the importance of background diet, a recent secondary analysis of the portfolio diet, a diet rich in fiber (e.g., barley, oats, psyllium, nuts) and soy products meant to lower cholesterol, was recently found to reduce BP similar to the DASH diet [128]. Lastly, high dietary K+ (associated with high fruit and vegetable consumption) is a hallmark of healthful diets and a meta-analysis of studies on K+ supplementation in patients without antihypertensive medication demonstrated that reducing Na+:K+ is associated with improved BP [129]. These findings on the importance of dietary K+ are in agreement with recent findings from PURE and TOHP in the general population [9•, 92].
Another consideration regarding background diet and salt include Na+ density in the diet. A recent secondary analysis of data obtained during the DASH diet trial demonstrated that dietary sodium density (mg Na+/energy consumed in kilocalories) was an important consideration regarding BP responses to the diet intervention [130••]. Interestingly, even on the control diet consisting of the widely recommended 2300 mg Na+/d, both systolic and diastolic BP were higher among those with lower energy intake (i.e., higher Na+ density) than among those with higher energy intake (i.e., lower Na+ density). These findings suggest that, rather than assigning an absolute Na+ load, recommendations should be based on energy intake. In fact, the original DASH trial sought to determine the influences of Na+ density (mg/kcal) at three levels, rather than specific absolute amounts [131]. This is an important consideration because Na+ and energy intake are highly correlated as nearly all segments of the US population consume ~ 1.7 mg Na+/kcal [1]. This is also important because, as highlighted in a recent AHA scientific statement on salt sensitivity of BP [77••], some studies have demonstrated females to be more salt-sensitive suggesting we may need to consider sex-specific dietary salt recommendations.
Conclusion
What has been established in the literature reviewed here is that dietary Na+ correlates with BP such that reductions in dietary Na+ lead to reductions in BP, a leading risk factor for CV disease. Additionally, high dietary salt contributes to target organ damage independent of BP. For example, high dietary salt has been shown to cause or be associated with systemic vascular dysfunction, arterial stiffening, altered renal function, left ventricular hypertrophy, skin Na+ deposition, cerebral circulatory dysfunction, alterations in sympathetic outflow, and potentially changes in bone content. Common underlying mechanisms include excess inflammation and oxidative stress. Despite the effects of high-salt diets on BP and target end organs, studies relating dietary salt consumption to CV events have produced controversial findings and this appears to be largely due to limitations related to difficulties in assessing habitual Na+ consumption. A major critique of studies finding that reduced salt is paradoxically related to increased CV events has been the lack of biological plausibility related to lower Na+ and adverse outcomes [103]. This criticism is supported by pre-clinical and clinical studies demonstrating that Na+ adversely affects several target organs, in some cases independent of BP (see Figure 1). Nonetheless, more data are needed to resolve the controversy. Lastly, there are data demonstrating that we should also consider sodium density in the diet rather than an absolute amount per se, and that background diet is important in mediating the CV consequences of dietary salt.
Abbreviations
- BP:
-
Blood pressure
- BMC:
-
Bone mineral content
- BMD:
-
Bone mineral density
- CV:
-
Cardiovascular
- CSF:
-
Cerebrospinal fluid
- CRIC :
-
Chronic Renal Insufficiency Cohort
- ENaC:
-
Epithelial sodium channel
- DASH:
-
Dietary approaches to stop hypertension
- LV:
-
Left ventricle
- NO:
-
Nitric oxide
- O2 - :
-
Superoxide
- ONOO- :
-
Peroxynitrate
- OVLT:
-
Organum vasculosum of the lamina terminalis
- PURE:
-
Prospective Urban Rural Epidemiology
- RCTs:
-
Randomized clinical trials
- RAAS:
-
Renin angiotensin aldosterone system
- RVLM:
-
Rostral ventrolateral medulla
- Na+ :
-
Sodium
- SOD:
-
Superoxide dismutase
- SNA:
-
Sympathetic nerve activity
- TOHP:
-
Trials of Hypertension Prevention
- VEGF:
-
Vascular endothelial growth factor
- 24hU-Na+ :
-
24-h urinary sodium excretion
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Quader Z, Zhao L, Gillespie C, Cogswell M, Terry A, Moshfegh A, et al. Sodium intake among persons aged ≥2 years — United States, 2013–2014. MMWR Morb Mortal Wkly. 2017;66(12):324–238 These are the most recent sodium consumption data published by the Centers for Disease Control and Prevention.
Bailey RL, Parker EA, Rhodes DG, Goldman JD, Clemens JC, Moshfegh AJ, et al. Estimating sodium and potassium intakes and their ratio in the American diet: data from the 2011-2012 NHANES. J Nutr. 2016.
Appel LJ, Frohlich ED, Hall JE, Pearson TA, Sacco RL, Seals DR, et al. The importance of population-wide sodium reduction as a means to prevent cardiovascular disease and stroke: a call to action from the American Heart Association. Circulation. 2011;123(10):1138–43.
Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344(1):3–10.
Cook NR, Appel LJ, Whelton PK. Lower levels of sodium intake and reduced cardiovascular risk. Circulation. 2014;129(9):981–9.
He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. Bmj. 2013;346:f1325.
Graudal NA, Hubeck-Graudal T, Jurgens G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev. 2017;4(Cd004022).
Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346.
• Mente A, O'Donnell M, Rangarajan S, McQueen M, Dagenais G, Wielgosz A, et al. Urinary sodium excretion, blood pressure, cardiovascular disease, and mortality: a community- level prospective epidemiological cohort study. Lancet. 2018;392(10146):496–506 The most recent dietary sodium study from the Prospective Urban Rural Epidemiology cohort. Although this study has been criticized for using spot urine samples to derive conclusions, sodium intake was associated with cardiovascular disease and strokes only in communities where mean intake was greater than 5000 mg/day. Overall, mean sodium intake and major cardiovascular events showed significant deviations from linearity due to a significant inverse association in the lowest tertile of sodium intake; thus, this study contributed to the controversy surround low sodium intake.
Mente A, O’Donnell M, Rangarajan S, Dagenais G, Lear S, McQueen M, et al. Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: a pooled analysis of data from four studies. Lancet. 2016;388(10043):465–75.
Stolarz-Skrzypek K, Kuznetsova T, Thijs L, Tikhonoff V, Seidlerova J, Richart T, et al. Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. Jama. 2011;305(17):1777–85.
O'Donnell M, Mente A, Yusuf S. Sodium intake and cardiovascular health. Circ Res. 2015;116(6):1046–57.
Harnack LJ, Cogswell ME, Shikany JM, Gardner CD, Gillespie C, Loria CM, et al. Sources of sodium in US adults from 3 geographic regions. Circulation. 2017;135(19):1775–83.
Lenda DM, Boegehold MA. Effect of a high-salt diet on oxidant enzyme activity in skeletal muscle microcirculation. Am J Physiol Heart Circ Physiol. 2002;282(2):H395–402.
Nurkiewicz TR, Boegehold MA. High salt intake reduces endothelium-dependent dilation of mouse arterioles via superoxide anion generated from nitric oxide synthase. Am J Phys Regul Integr Comp Phys. 2007;292(4):R1550–6.
•• Guers JJ, Kasecky-Lardner L, Farquhar WB, Edwards DG, Lennon SL. Voluntary wheel running prevents salt-induced endothelial dysfunction: role of oxidative stress. J Appl Physiol. 1985;2018 Our group recently presented data suggesting that voluntary wheel running can prevent impairments in endothelium-dependent relaxation in the femoral artery of rats fed a high-salt diet. These findings appear to be independent of blood pressure and mediated through a decrease in expression of NADPH oxidases and an increase of superoxide dismutase as a result of physical activity.
Matthews EL, Brian MS, Ramick MG, Lennon-Edwards S, Edwards DG, Farquhar WB. High dietary sodium reduces brachial artery flow-mediated dilation in humans with salt-sensitive and salt-resistant blood pressure. J Appl Physiol. 2015;118(12):1510–5.
Lennon-Edwards S, Ramick MG, Matthews EL, Brian MS, Farquhar WB, Edwards DG. Salt loading has a more deleterious effect on flow-mediated dilation in salt-resistant men than women. Nutr Metab Cardiovasc Dis. 2014;24(9):990–5.
Tzemos N, Lim PO, Wong S, Struthers AD, MacDonald TM. Adverse cardiovascular effects of acute salt loading in young normotensive individuals. Hypertension. 2008;51(6):1525–30.
Jablonski KL, Racine ML, Geolfos CJ, Gates PE, Chonchol M, McQueen MB, et al. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J Am Coll Cardiol. 2013;61(3):335–43.
Muth BJ, Brian MS, Chirinos JA, Lennon SL, Farquhar WB, Edwards DG. Central systolic blood pressure and aortic stiffness response to dietary sodium in young and middle-aged adults. J Am Soc Hypertens. 2017;11(10):627–34.
Dickinson KM, Clifton PM, Keogh JB. A reduction of 3 g/day from a usual 9 g/day salt diet improves endothelial function and decreases endothelin-1 in a randomised cross_over study in normotensive overweight and obese subjects. Atherosclerosis. 2014;233(1):32–8.
Eisenach JH, Gullixson LR, Kost SL, Joyner MJ, Turner ST, Nicholson WT. Sex differences in salt sensitivity to nitric oxide dependent vasodilation in healthy young adults. J Appl Physiol (1985). 2012;112(6):1049–53.
Greaney JL, DuPont JJ, Lennon-Edwards SL, Sanders PW, Edwards DG, Farquhar WB. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: role of oxidative stress. J Physiol. 2012;590(21):5519–28.
Cavka A, Jukic I, Ali M, Goslawski M, Bian JT, Wang E, et al. Short-term high salt intake reduces brachial artery and microvascular function in the absence of changes in blood pressure. J Hypertens. 2016;34(4):676–84.
D'Elia L, Galletti F, La Fata E, Sabino P, Strazzullo P. Effect of dietary sodium restriction on arterial stiffness: systematic review and meta-analysis of the randomized controlled trials. J Hypertens. 2018;36(4):734–43.
Dickinson KM, Clifton PM, Burrell LM, Barrett PH, Keogh JB. Postprandial effects of a high salt meal on serum sodium, arterial stiffness, markers of nitric oxide production and markers of endothelial function. Atherosclerosis. 2014;232(1):211–6.
Dickinson KM, Clifton PM, Keogh JB. Endothelial function is impaired after a high-salt meal in healthy subjects. Am J Clin Nutr. 2011;93(3):500–5.
Lenda DM, Sauls BA, Boegehold MA. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol. 2000;279(1):H7–h14.
Edwards DG, Farquhar WB. Vascular effects of dietary salt. Curr Opin Nephrol Hypertens. 2015;24(1):8–13.
Sessa WC. eNOS at a glance. J Cell Sci. 2004;117(Pt 12):2427–9.
Wray DW, Witman MA, Ives SJ, McDaniel J, Trinity JD, Conklin JD, et al. Does brachial artery flow-mediated vasodilation provide a bioassay for NO? Hypertension. 2013;62(2):345–51.
Loscalzo J. The identification of nitric oxide as endothelium-derived relaxing factor. Circ Res. 2013;113(2):100–3.
Durand MJ, Lombard JH. Low-dose angiotensin II infusion restores vascular function in cerebral arteries of high salt–fed rats by increasing copper/zinc superoxide dimutase expression. Am J Hypertens. 2013;26(6):739–47.
Martin JV, Liberati DM, Diebel LN. Excess sodium is deleterious on endothelial and glycocalyx barrier function: a microfluidic study. J Trauma Acute Care Surg. 2018;85(1):128–34.
Tasevska I, Enhörning S, Burri P, Melander O. High salt intake increases copeptin but salt sensitivity is associated with fluid induced reduction of copeptin in women. Int J Hypertens. 2014;2014:1–5.
Spinelli L, Golino P, Piscione F, Chiariello M, Focaccio A, Ambrosio G, et al. Effects of oral salt load on arginine-vasopressin secretion in normal subjects. Ann Clin Lab Sci. 1987;17(5):350–7.
Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration- dependent signaling pathways. J Appl Physiol. 2007;102(4):1402–9.
Vranish JR, Holwerda SW, Young BE, Credeur DP, Patik JC, Barbosa TC, et al. Exaggerated vasoconstriction to spontaneous bursts of muscle sympathetic nerve activity in healthy young black men. Hypertension. 2018;71(1):192–8.
•• Babcock MC, Robinson AT, Migdal KU, Watso JC, Wenner MM, Stocker SD, et al. Reducing dietary sodium to 1000 mg per day reduces neurovascular transduction without stimulating sympathetic outflow. Hypertension. 2019;73(3):587–93 We demonstrated that low (1000 mg/day) vs recommended (2300 mg/day) sodium does not influence resting sympathetic nerve activity but the low-sodium diet did reduce sympathetic vascular transduction.
• Hall JE. Kidney dysfunction mediates salt-induced increases in blood pressure. Circulation. 2016;133(9):894–906 Kidney dysfunction, characterized by impaired pressure natriuresis, has been demonstrated in experimental models and human salt-sensitive hypertension. Mechanisms contributing to abnormalities of kidney function that lead to increased NaCl reabsorption decreased glomerular capillary filtration coefficient or cause nephron injury/loss are discussed in this review.
•• Wenner MM, Paul EP, Robinson AT, Rose WC, Farquhar WB. Acute NaCl loading reveals a higher blood pressure for a given serum sodium level in black compared to white adults. Front Physiol. 2018;9 We recently demonstrated black individuals exhibit augmented serum sodium to an acute hypertonic saline load and greater blood pressure responsiveness to a given serum sodium.
Pavlov TS, Staruschenko A. Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Ren Physiol. 2017;313(2):F135–f40.
Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24(7):1053–62.
Chakraborty S, Galla S, Cheng X, Yeo JY, Mell B, Singh V, et al. Salt-responsive metabolite, beta-hydroxybutyrate, attenuates hypertension. Cell Rep. 2018;25(3):677–89.e4.
Farquhar WB, Edwards DG, Jurkovitz CT, Weintraub WS. Dietary sodium and health: more than just blood pressure. J Am Coll Cardiol. 2015;65(10):1042–50.
Frame AA, Wainford RD. Mechanisms of altered renal sodium handling in age-related hypertension. Am J Physiol Ren Physiol. 2018;315(1):F1–f6.
Gomes AC, Falcao-Pires I, Pires AL, Bras-Silva C, Leite-Moreira AF. Rodent models of heart failure: an updated review. Heart Fail Rev. 2013;18(2):219–49.
van der Westhuizen B, Schutte AE, Gafane-Matemane LF, Kruger R. Left ventricular mass independently associates with 24-hour sodium excretion in young masked hypertensive adults: the African-PREDICT study. Int J Cardiol. 2019;276:218–23.
Aronow WS. Hypertension and left ventricular hypertrophy. Ann Transl Med. 2017;5(15).
Jin Y, Kuznetsova T, Maillard M, Richart T, Thijs L, Bochud M, et al. Independent relations of left ventricular structure with the 24-hour urinary excretion of sodium and aldosterone. Hypertension. 2009;54(3):489–95.
du Cailar G, Fesler P, Ribstein J, Mimran A. Dietary sodium, aldosterone, and left ventricular mass changes during long-term inhibition of the renin-angiotensin system. Hypertension. 2010;56(5):865–70.
Haring B, Wang W, Lee ET, Jhamnani S, Howard BV, Devereux RB. Effect of dietary sodium and potassium intake on left ventricular diastolic function and mass in adults</=40 years (from the Strong Heart Study). Am J Cardiol. 2015;115(9):1244–8.
Cobb LK, Anderson CA, Elliott P, Hu FB, Liu K, Neaton JD, et al. Methodological issues in cohort studies that relate sodium intake to cardiovascular disease outcomes: a science advisory from the American Heart Association. Circulation. 2014;129(10):1173–86.
•• Selvarajah V, Connolly K, McEniery C, Wilkinson I. Skin sodium and hypertension: a paradigm shift? Curr Hypertens Rep. 2018;20(11) One of two excellent recent reviews that summarize studies demonstrating observations of sodium storage in the skin as a means to buffer free extracellular Na+. Also discussed is the role downstream of macrophage modulation of the extracellular matrix and lymphatics network, suggesting complex extrarenal regulatory mechanisms that contribute to electrolyte homeostasis in the body.
•• Wiig H, Luft FC, Titze JM. The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol (Oxford). 2018;222(3) Second of two excellent recent reviews that summarize studies demonstrating observations of sodium storage in the skin as a means to buffer free extracellular Na+. Also discussed is the role downstream of macrophage modulation of the extracellular matrix and lymphatics network, suggesting complex extrarenal regulatory mechanisms that contribute to electrolyte homeostasis in the body.
Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–52.
Machnik A, Dahlmann A, Kopp C, Goss J, Wagner H, van Rooijen N, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. 2010;55(3):755–61.
Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127(5):1944–59.
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–22.
Foss JD, Kirabo A, Harrison DG. Do high-salt microenvironments drive hypertensive inflammation? Am J Phys Regul Integr Comp Phys. 2017;312:R1–4.
Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Muller DN, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61(3):635–40.
Kopp C, Linz P, Maier C, Wabel P, Hammon M, Nagel AM, et al. Elevated tissue sodium deposition in patients with type 2 diabetes on hemodialysis detected by (23) Na magnetic resonance imaging. Kidney Int. 2018;93(5):1191–7.
Crescenzi R, Marton A, Donahue PMC, Mahany HB, Lants SK, Wang P, et al. Tissue sodium content is elevated in the skin and subcutaneous adipose tissue in women with lipedema. Obesity (Silver Spring). 2018;26(2):310–7.
Schneider MP, Raff U, Kopp C, Scheppach JB, Toncar S, Wanner C, et al. Skin sodium concentration correlates with left ventricular hypertrophy in CKD. J Am Soc Nephrol. 2017;28(6):1867–76.
•• Selvarajah V, Maki-Petaja KM, Pedro L, SFA B, Burling K, Goodhart AK, et al. Novel mechanism for buffering dietary salt in humans: effects of salt loading on skin sodium, vascular endothelial growth factor c, and blood pressure. Hypertension. 2017;70(5):930–7 The findings of this study suggest that the skin sodium deposition may buffer dietary sodium; however, this relation between dietary sodium and skin sodium may be influenced by sex. Only male participants exhibited an increase in skin sodium following a high-salt diet.
Perry IJ, Beevers DG. Salt intake and stroke: a possible direct effect. J Hum Hypertens. 1992;6(1):23–5.
Nagata C, Takatsuka N, Shimizu N, Shimizu H. Sodium intake and risk of death from stroke in Japanese men and women. Stroke. 2004;35(7):1543–7.
Cosic A, Jukic I, Stupin A, Mihalj M, Mihaljevic Z, Novak S, et al. Attenuated flow- induced dilatation of middle cerebral arteries is related to increased vascular oxidative stress in rats on a short-term high salt diet. J Physiol. 2016;594(17):4917–31.
Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018;21(2):240–9.
Allen LA, Schmidt JR, Thompson CT, Carlson BE, Beard DA, Lombard JH. High salt diet impairs cerebral blood flow regulation via salt-induced angiotensin II suppression. Microcirculation. 2018;e12518.
Markus H, Cullinane M. Severely impaired cerebrovascular reactivity predicts stroke and TIA risk in patients with carotid artery stenosis and occlusion. Brain. 2001;124(Pt 3):457–67.
Fisher JP, Young CN, Fadel PJ. Central sympathetic overactivity: maladies and mechanisms. Auton Neurosci. 2009;148(1–2):5–15.
Babcock MC, Brian MS, Watso JC, Edwards DG, Stocker SD, Wenner MM, et al. Alterations in dietary sodium intake affect cardiovagal baroreflex sensitivity. Am J Phys Regul Integr Comp Phys. 2018;315(4):R688–R95.
Stocker SD, Lang SM, Simmonds SS, Wenner MM, Farquhar WB. Cerebrospinal fluid hypernatremia elevates sympathetic nerve activity and blood pressure via the rostral ventrolateral medulla. Hypertension. 2015;66(6):1184–90.
Kawano Y, Yoshida K, Kawamura M, Yoshimi H, Ashida T, Abe H, et al. Sodium and noradrenaline in cerebrospinal fluid and blood in salt-sensitive and non-salt-sensitive essential hypertension. Clin Exp Pharmacol Physiol. 1992;19(4):235–41.
•• Elijovich F, Weinberger MH, Anderson CA, Appel LJ, Bursztyn M, Cook NR, et al. Salt sensitivity of blood pressure: a scientific statement from the American Heart Association. Hypertension. 2016;68(3):e7-e46 Salt sensitivity of blood pressure is a phenomenon whereby some members of the population exhibit blood pressure changes that parallel changes in salt intake. This recent scientific statement from the American Heart Association summarizes the prevalence and some of the mechanisms that contribute to salt sensitivity of blood pressure.
Kinsman BJ, Simmonds SS, Browning KN, Stocker SD. Organum vasculosum of the lamina terminalis detects NaCl to elevate sympathetic nerve activity and blood pressure. Hypertension. 2017;69(1):163–70.
Kinsman BJ, Browning KN, Stocker SD. NaCl and osmolarity produce different responses in organum vasculosum of the lamina terminalis neurons, sympathetic nerve activity and blood pressure. J Physiol. 2017;595(18):6187–201.
Kinsman BJ, Nation HN, Stocker SD. Hypothalamic signaling in body fluid homeostasis and hypertension. Curr Hypertens Rep. 2017;19(6):50.
Duvernoy HM, Risold PY. The circumventricular organs: an atlas of comparative anatomy and vascularization. Brain Res Rev. 2007;56(1):119–47.
Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335–46.
Badoer E. Hypothalamic paraventricular nucleus and cardiovascular regulation. Clin Exp Pharmacol Physiol. 2001;28(1–2):95–9.
•• Nomura K, Hiyama TY, Sakuta H, Matsuda T, Lin CH, Kobayashi K, et al. [Na(+)] increases in body fluids sensed by central nax induce sympathetically mediated blood pressure elevations via H(+)-dependent activation of ASIC1a. Neuron. 2019;101(1):60–75.e6 This recent study provides insight into the neurogenic mechanisms responsible for salt-induced blood pressure elevations. Specifically, it was demonstrated that Nax channels are expressed in specific glial cells in the organum vasculosum lamina terminalis and act as sensors detecting increases in sodium in body fluids. The organum vasculosum lamina terminalis neurons projecting to the paraventricular nucleus are activated via acid-sensing ion channel 1a (ASIC1a) by hydrogen ions released from Nax-positive glial cells.
Kim SW, Jeon JH, Choi YK, Lee WK, Hwang IR, Kim JG, et al. Association of urinary sodium/creatinine ratio with bone mineral density in postmenopausal women: KNHANES 2008–2011. Endocrine. 2015;49(3):791–9.
Park Y, Kwon SJ, Ha YC. Association between urinary sodium excretion and bone health in male and female adults. Ann Nutr Metab. 2016;68(3):189–96.
Cao WT, He J, Chen GD, Wang C, Qiu R, Chen YM. The association between urinary sodium to potassium ratio and bone density in middle-aged Chinese adults. Osteoporos Int. 2017;28(3):1077–86.
Fatahi S, Namazi N, Larijani B, Azadbakht L. The association of dietary and urinary sodium with bone mineral density and risk of osteoporosis: a systematic review and meta- analysis. J Am Coll Nutr. 2018;37(6):522–32.
Carbone L, Johnson KC, Huang Y, Pettinger M, Thomas F, Cauley J, et al. Sodium intake and osteoporosis. Findings from the Women’s Health Initiative. J Clin Endocrinol Metab. 2016;101(4):1414–21.
Mozaffarian D, Fahimi S, Singh GM, Micha R, Khatibzadeh S, Engell RE, et al. Global sodium consumption and death from cardiovascular causes. N Engl J Med. 2014;371(7):624–34.
Mancia G, Oparil S, Whelton PK, McKee M, Dominiczak A, Luft FC, et al. The technical report on sodium intake and cardiovascular disease in low- and middle-income countries by the joint working group of the World Heart Federation, the European Society of Hypertension and the European Public Health Association. Eur Heart J. 2017;38(10):712–9.
Cook NR, Appel LJ, Whelton PK. Sodium intake and all-cause mortality over 20 years in the Trials of Hypertension Prevention. J Am Coll Cardiol. 2016;68(15):1609–17.
Mills KT, Chen J, Yang W, Appel LJ, Kusek JW, Alper A, et al. Sodium excretion and the risk of cardiovascular disease in patients with chronic kidney disease. Jama. 2016;315(20):2200–10.
Thomas MC, Moran J, Forsblom C, Harjutsalo V, Thorn L, Ahola A, et al. The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care. 2011;34(4):861–6.
Ekinci EI, Clarke S, Thomas MC, Moran JL, Cheong K, MacIsaac RJ, et al. Dietary salt intake and mortality in patients with type 2 diabetes. Diabetes Care. 2011;34(3):703–9.
Mente A, O'Donnell M, Rangarajan S, McQueen M, Dagenais G, Wielgosz A, et al. Urinary sodium excretion, blood pressure, cardiovascular disease, and mortality: a community- level prospective epidemiological cohort study. Lancet. 2018;392(10146):496–506.
O'Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med. 2014;371(7):612–23.
Graudal NA, Hubeck-Graudal T, Jurgens G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev. 2011;(11):Cd004022.
Lerchl K, Rakova N, Dahlmann A, Rauh M, Goller U, Basner M, et al. Agreement between 24-hour salt ingestion and sodium excretion in a controlled environment. Hypertension. 2015;66(4):850–7.
Allen NB, Zhao L, Loria CM, Van Horn L, Wang CY, Pfeiffer CM, et al. The validity of predictive equations to estimate 24-hour sodium excretion: the MESA and CARDIA Urinary Sodium Study. Am J Epidemiol. 2017;186(2):149–59.
Cogswell ME, Wang CY, Chen TC, Pfeiffer CM, Elliott P, Gillespie CD, et al. Validity of predictive equations for 24-h urinary sodium excretion in adults aged 18-39 y. Am J Clin Nutr. 2013;98(6):1502–13.
Tan M, He FJ, MacGregor GA. Salt and cardiovascular disease in PURE: a large sample size cannot make up for erroneous estimations. J Renin-Angiotensin-Aldosterone Syst. 2018;19(4):1470320318810015.
Cappuccio FP, Beer M, Strazzullo P. Population dietary salt reduction and the risk of cardiovascular disease. A scientific statement from the European Salt Action Network. Nutr Metab Cardiovasc Dis. 2018.
Graudal N, Jurgens G, Baslund B, Alderman MH. Compared with usual sodium intake, low- and excessive-sodium diets are associated with increased mortality: a meta-analysis. Am J Hypertens. 2014;27(9):1129–37.
•• Jones DW, Luft FC, Whelton PK, Alderman MH, Hall JE, Peterson ED, et al. Can we end the salt wars with a randomized clinical trial in a controlled environment? Hypertension. 2018;72(1):10–1 This editorial served as a call for dietary sodium outcomes clinical trial given the controversy between low sodium intake and cardiovascular outcomes caused by findings from a number of epidemiological studies.
The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in overweight people with high-normal blood pressure. The Trials of Hypertension Prevention, phase II. Arch Intern Med. 1997;157(6):657–67.
Byrd K, Almanza B, Ghiselli RF, Behnke C, Eicher-Miller HA. Reported action to decrease Sodium intake is associated with dining out frequency and use of menu nutrition information among US adults. J Acad Nutr Diet. 2018;118(5):824–35.
• Auchincloss AH, Leonberg BL, Glanz K, Bellitz S, Ricchezza A, Jervis A. Nutritional value of meals at full-service restaurant chains. J Nutr Educ Behav. 2014;46(1):75–81 This study determined the nutritional value of meals at full-service restaurant chains. Adult meals (defined as entrée + side dish + one-half appetizer) approximated ~ 3,500 mg sodium.
• McCallum L, Lip S, Padmanabhan S. The hidden hand of chloride in hypertension. Pflugers Arch. 2015;467(3):595–603 There is consensus that high sodium chloride intake increases blood pressure. This review summarizes the evidence supporting an independent role for chloride on hypertension and cardiovascular health.
Kurtz TW, Al-Bander HA, Morris RC Jr. "salt-sensitive" essential hypertension in men. Is the sodium ion alone important? N Engl J Med. 1987;317(17):1043–8.
Shore AC, Markandu ND, MacGregor GA. A randomized crossover study to compare the blood pressure response to sodium loading with and without chloride in patients with essential hypertension. J Hypertens. 1988;6(8):613–7.
Schorr U, Distler A, Sharma AM. Effect of sodium chloride- and sodium bicarbonate-rich mineral water on blood pressure and metabolic parameters in elderly normotensive individuals: a randomized double-blind crossover trial. J Hypertens. 1996;14(1):131–5.
Luft FC, Zemel MB, Sowers JA, Fineberg NS, Weinberger MH. Sodium bicarbonate and sodium chloride: effects on blood pressure and electrolyte homeostasis in normal and hypertensive man. J Hypertens. 1990;8(7):663–70.
Oh SW, Koo HS, Han KH, Han SY, Chin HJ. Associations of sodium intake with obesity, metabolic disorder, and albuminuria according to age. PLoS One. 2017;12.
Monteiro CA, Moubarac JC, Levy RB, Canella DS, Louzada M, Cannon G. Household availability of ultra-processed foods and obesity in nineteen European countries. Public Health Nutr. 2018;21(1):18–26.
Costa CS, Rauber F, Leffa PS, Sangalli CN, Campagnolo PDB, Vitolo MR. Ultra- processed food consumption and its effects on anthropometric and glucose profile: a longitudinal study during childhood. Nutr Metab Cardiovasc Dis. 2019;29(2):177–84.
Bolhuis DP, Costanzo A, Newman LP, Keast RS. Salt promotes passive overconsumption of dietary fat in humans. J Nutr. 2016;146(4):838–45.
Zhang Y, Li F, Liu FQ, Chu C, Wang Y, Wang D, et al. Elevation of fasting ghrelin in healthy human subjects consuming a high-salt diet: a novel mechanism of obesity? Nutrients. 2016;8(6).
Zhao L, Cogswell ME, Yang Q, Zhang Z, Onufrak S, Jackson SL, et al. Association of usual 24-h sodium excretion with measures of adiposity among adults in the United States: NHANES, 2014. Am J Clin Nutr. 2019;109(1):139–47.
Juraschek SP, Miller ER 3rd, Weaver CM, Appel LJ. Effects of sodium reduction and the DASH diet in relation to baseline blood pressure. J Am Coll Cardiol. 2017;70(23):2841–8.
Fulgoni VL, Agarwal S, Spence L, Samuel P. Sodium intake in US ethnic subgroups and potential impact of a new sodium reduction technology: NHANES dietary modeling. Nutr J. 2014;13.
Dubowitz T, Heron M, Bird CE, Lurie N, Finch BK, Basurto-Dávila R, et al. Neighborhood socioeconomic status and fruit and vegetable intake among whites, blacks, and Mexican- Americans in the United States. Am J Clin Nutr. 2008;87(6):1883–91.
Fahlman MM, McCaughtry N, Martin J, Shen B. Racial and socioeconomic disparities in nutrition behaviors: targeted interventions needed. J Nutr Educ Behav. 2010;42(1):10–6.
Wang Y, Chen X. How much of racial/ethnic disparities in dietary intakes, exercise, and weight status can be explained by nutrition- and health-related psychosocial factors and socioeconomic status among US adults? J Am Diet Assoc. 2011;111(12):1904–11.
Storey M, Anderson P. Income and race/ethnicity influence dietary fiber intake and vegetable consumption. Nutr Res (New York, NY). 2014;34(10):844–50.
Nicklett EJ, Kadell AR. Fruit and vegetable intake among older adults: a scoping review. Maturitas. 2013;75(4):305–12.
Kirkpatrick SI, Dodd KW, Reedy J, Krebs-Smith SM. Income and race/ethnicity are associated with adherence to food-based dietary guidance among US adults and children. J Acad Nutr Diet. 2012;112(5):624–35.e6.
Jenkins DJ, Jones PJ, Frohlich J, Lamarche B, Ireland C, Nishi SK, et al. The effect of a dietary portfolio compared to a DASH-type diet on blood pressure. Nutr Metab Cardiovasc Dis. 2015;25(12):1132–9.
Binia A, Jaeger J, Hu Y, Singh A, Zimmermann D. Daily potassium intake and sodium-to- potassium ratio in the reduction of blood pressure: a meta-analysis of randomized controlled trials. J Hypertens. 2015;33(8):1509–20.
•• Murtaugh MA, Beasley JM, Appel LJ, Guenther PM, McFadden M, Greene T, et al. Relationship of sodium intake and blood pressure varies with energy intake: secondary analysis of the DASH (Dietary Approaches to Stop Hypertension)-Sodium Trial. Hypertension. 2018;71(5):858–65 Dietary sodium recommendations are expressed as absolute amounts (mg/day) rather than in sodium density (mg/kcal); however, this manuscript determined there is a relation between sodium density and blood pressure. Additionally, it is difficult for larger individuals to consume lower amounts of sodium and smaller individuals may not experience benefits with the current absolute sodium recommendations. Taken together, these data suggest more research is needed to confirm if recommendations should be normalized body weight or caloric intake.
Svetkey LP, Sacks FM, Obarzanek E, Vollmer WM, Appel LJ, Lin PH, et al. The DASH diet, sodium intake and blood pressure trial (DASH-sodium): rationale and design. DASH- Sodium Collaborative Research Group. J Am Diet Assoc. 1999;99(8 Suppl):S96–104.
Funding
The authors have been supported by the following grants which have also contributed to some of the original work cited here: NIH P20 GM113125 (DGE); NIH R01 HL128388 (WBF); NIH R01 HL104106 (WBF and DGE); AHA 18POST34060020 (ATR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Blood Pressure Monitoring and Management
Rights and permissions
About this article

Cite this article
Robinson, A.T., Edwards, D.G. & Farquhar, W.B. The Influence of Dietary Salt Beyond Blood Pressure. Curr Hypertens Rep 21, 42 (2019). https://doi.org/10.1007/s11906-019-0948-5
Published:
DOI: https://doi.org/10.1007/s11906-019-0948-5