
Abstract
Purpose of Review
The discovery of innate lymphoid cells (ILCs) over the past decade has reformed principles that were once thought to be exclusive to adaptive immunity. Here, we describe ILC nomenclature and function, and provide a survey of studies examining these cells in the context of HIV/SIV infections. Particular emphasis is placed on the ILC3 subset, important for proper functioning of the gastrointestinal tract barrier.
Recent Findings
Studies in both humans and nonhuman primates have found ILCs to be rapidly and durably depleted in untreated HIV/SIV infections. Their depletion is most likely due to a number of bystander effects induced by viral replication.
Summary
Given the number of associations observed between loss of ILCs and HIV-related GI damage, their impact on the GI tract is likely important. It may be informative to examine this subset in parallel with other immune cell types when assessing overall health of the GI tract in future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Much of our understanding of the human immune system has been based upon measurements made from peripheral blood. Importantly, analysis of peripheral blood is often a poor surrogate for immune activity in tissues, where the majority of leukocytes reside. Indeed, secondary lymphoid tissues serve as sites for adaptive immune cell priming and support a homeostatic niche for lymphocytes. Moreover, the gastrointestinal (GI) tract promotes T and B cell tolerance to commensal bacterial antigens and provides protection against pathogens. The net interactions among microbiota, leukocytes, and nonleukocytes support the structural integrity of tissues, and dysfunctions among immune cell populations oftentimes correspond to an architectural breakdown of the tissue that harbors those cells. The pathogenesis of HIV-1 infection can, in many respects, be explained by a breakdown of the immune and structural components of tissues. For example, there is now overwhelming evidence that CD4 T cells in the GI tract are depleted very early in HIV disease course [1,2,3]. Loss of these cells disrupts the structural barrier of the GI tract, allowing microbial products to spread systemically within the host and perpetuate immune activation [4]. Lymphoid tissues are also a prominent site of pathogenesis. These are sites of viral replication and persistence and become fibrotic in HIV-1 infection, depriving naïve T cells of essential signals needed for their survival [5, 6]. In each of these instances, suppression of HIV replication with combined antiretroviral therapy (cART) does not completely normalize GI or lymphoid tissue damage sustained in untreated infection [7]. There are thus a number of therapeutic modalities for supplementation of cART aimed at reversing HIV-associated tissue damage [8, 9]. Recently, innate lymphoid cells (ILCs) have emerged as critical regulators of tissue homeostasis. While ILCs constitute only a small fraction of the hematopoietic cell pool, they share many functional similarities to adaptive immune cells and have been shown to be of critical importance. They exert their functions in response to innate stimuli (rather than antigen recognition), which allows the ILC population to provide a source of effector cytokines at more rapid kinetics than the adaptive immune cells. A growing number of reports indicate that GI damage in HIV and SIV infection is associated with rapid depletion and functional alterations to ILCs. Here, we introduce overarching features of ILCs, with particular focus on the ILC3 subset and its role in GI tissue homeostasis. We then discuss how these cells become perturbed in HIV/SIV infections and potential mechanisms for these dysfunctions.
ILC Nomenclature
The existence of ILCs that have functionalities similar to CD4 T helper cells was first reported in 2008, when a natural killer (NK)-like cell population present in human mucosal-associated lymphoid tissues which secreted IL-22 was described [10]. Shortly thereafter, a population of c-Kit+ cells capable of secreting large amounts of IL-13 was described in mice [11,12,13]. At the time, these were coined NK-22, natural helper cells, or nuocytes. These cells are now defined as ILCs. ILCs possess a lymphoid morphology yet lack genetically rearranged antigen receptors or lineage markers that define other immune cell types, including CD3 (T cells), CD11c (mDCs), CD123 (pDCs), CD16 (NK cells), CD20/CD19 (B cells), CD23 (granulocytes), and CD34 (stem cells) [14]. These cells express high levels of γc chain cytokine receptors and in doing so influence the availability of γ-chain cytokines in primary and secondary lymphoid tissues [15]. ILCs can currently be classified into one of three ILC populations—ILC1, ILC2, and ILC3—based upon expression of cytokine and lineage-defining transcription factors, which largely mirror classification of Th1, Th2, and Th17 CD4+ T cells [14].
ILC1s
ILC1s are classified by their expression of the T-box transcription factor (T-bet) and their functional ability to secrete IFNγ in response to IL-12 and IL-18 [16]. They are defined as Lineage−CD127+, and lack the prostaglandin DP2 receptor (CRTH2) and stem cell growth factor receptor (c-Kit) surface markers that define ILC2 and ILC3 subtypes, respectively. Defining features of ILC1s are thought to include their permanent tissue residency, uniform CD127 expression, and generally lower degree of cytotoxic potential in comparison to their classical NK cell counterparts [17,18,19].
While some distinctions have been noted, ILC1s overlap considerably in phenotype and function to classical NK cells. For example, while CD127 is expressed on all ILC1s, human CD16−CD56bright NK cells (capable of secreting IFNγ) also express high surface levels of CD127 [20]. Moreover, T-bet, the lineage-defining transcription factor of ILC1s, has been reported to be preferentially expressed in ILC1s of some human studies, but not others [16, 21••, 22••, 23]. The difficulties in drawing interpretations from this population may lie in a current lack of surface markers that can define ILC1s unambiguously. Indeed, single-cell analysis of human tonsillar ILC1s defined as Lineage−CD127+CRTH2−c-Kit− has found this population to be enriched for transcripts encoding TCR variable regions and other T cell-specific markers including CD4, CD5, and CD28 [23]. This is in line with other reports describing a putative “CD4+ ILC1” [24, 25]. An additional study has found that, unlike ILC2s and ILC3s in tonsils, ILC1s do not form distinct clusters by 2-dimensional t-SNE plots, and instead overlap with T cell, dendritic cell, and ILC3 clusters [21••]. Thus, the existence of ILC1s in primates is not entirely clear. However, in vitro expansion of human ILC precursors has identified clones that differentiate into T-bet+ IFNγ-secreting cells which are distinct from Eomes+ CD94+ NK-like clones [26, 27]. At the very least, it is reasonable to say that the field would clearly benefit from the identification of an ILC1-specific marker, yet, at present, interpretations drawn from this population as defined currently should be done with caution given its apparent heterogeneity.
ILC2s
ILC2s are defined by their high expression of the GATA-3 transcription factor and potential to secrete the Th2-related cytokines IL-4, IL-5, and IL-13 in response to IL-25, TSLP, or IL-33 [11, 28, 29]. They also express CRTH2 and CD161 [30]. ST2, the IL-33 receptor, can also be used to define these cells although the expression of this marker on ILC2s across all anatomical sites is not uniform (i.e., blood CRTH2+ ILC2s lack ST2 expression) [31].
Similar to T-helper subsets, ILC2s can alter their phenotype and function when exposed to particular polarizing signals. For example, ILC2s can downregulate surface expression of ST2 and convert to IFNγ-producing ILC1-like cells both in vitro and in vivo [31,32,33]. The presence of IL-12 and the alarmin IL-25, with upregulation of the Th1-associated transcription factor T-bet, are important for this process [34].
ILC2s have been classically implicated in their defense against helminthic infections, where Th2-related cytokines are important for parasitic expulsion [35]. However, their influence in health and disease has proved to be surprisingly more broad, where they contribute to regulating eosinophil homeostasis and beiging of white adipose tissue at steady-state conditions, and allergic airway inflammation during inflammatory conditions [36,37,38]. To date, there is not a great deal literature of these cells in contexts of viral infections or GI barrier damage (classically implicated by Th1 or Th17-type responses, respectively). ILC2 biology is covered more in-depth in other reviews [39, 40].
ILC3s
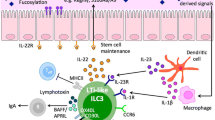
More heterogenous than ILC2s, the ILC3 subtype is defined by expression of RAR-related orphan receptor gamma (RORγt), with potential to secrete GM-CSF, IL-17, and IL-22 in response to IL-1β and IL-23, and surface expression of c-Kit [10, 41, 42]. They can be divided into at least three distinct cell types: LTi cells, natural cytotoxicity receptor (NCR) −, and NCR+ ILC3s. NKp46 and NKp44 mark NCR+ ILC3s in mice and humans, respectively, and it is clear this marker imparts a distinct functional state that is more activated and polyfunctional than the NCR− ILC3 subset [23, 43]. Like the ILC2s, ILC3s can become more “ILC1-like” in phenotype and function, losing RORγt and concomitantly upregulating T-bet when exposed to IL-12, IL-15, and IL1β [44, 45]. ILC3s exert their effector functions primarily in gut mucosal tissues, where they are particularly enriched compared to other anatomical sites of the body [21••, 46••].
ILC3s in GI Tissue Homeostasis
The importance of ILC3s in the anatomy and physiology of the GI tract is highlighted by the fact that depletion of these cells in RAG−/− mice leads to peripheral dissemination of commensal bacteria and systemic inflammation [47]. This phenotype can be rescued by exogenous IL-22 administration, underscoring a critical mechanism by which ILC3s promote intestinal homeostasis. Indeed, IL-22 (and also IL-17) can act directly on GI tract epithelial cells to proliferate and to enhance expression of defensins, mucins, tight junctions, and antimicrobial peptides that both strengthen the GI barrier and provide an essential line of defense against bacterial pathogens [48,49,50,51,52]. Given the key role that IL-17 and IL-22 secretion play in ILC3 effector function, many groups have sought to understand the external cues within the GI microenvironment that influence this process. While ILCs are innate immune cells, ILC3-derived secretion of IL-22 is likely not Toll-like receptor (TLR)-dependent [53]. Instead, there is growing evidence that myeloid cells in the gut provide key external cues that regulate functionality of ILC3s. The interactions between these two cell types are likely twofold: (1) myeloid-derived interactions provide an important source of IL-23 to ILC3s (a potent inducer of IL-22); and (2) myeloid cells can serve as an intermediary regulator of ILC3 interactions with the microbiota. For example, in mice, CX3CR1+ mononuclear phagocytes are found to be in close proximity to ILC3s and can support ILC3-derived IL-22 production in an IL-23- and IL-1β-dependent manner [54]. CCR2+ DCs in the gut can also be a source of IL-23, and their impact on IL-22 production in ILC3s can be abolished with broad-spectrum antibiotics, or subsequently rescued with segmented filamentous bacterial colonization [55].
While these studies implicate critical roles for myeloid cells in ILC3 interactions with the microbiota, there are nevertheless ways in which the microbiota can regulate ILC3 effector function directly. One of which is through microbial metabolites. Butyric acid, a short-chain fatty acid, can negatively regulate IL-22 production in ILC3s [56]. Moreover, both tryptophan catabolites and microbial metabolic products are ligands for the aryl hydrocarbon receptor [57,58,59], a transcription factor shown to be essential for maintaining IL-22 production in ILC3s [60,61,62]. Thus, multiple regulatory mechanisms exist, both microbial and host-derived, that serve to promote healthy functioning of the gut through ILC3 effector function.
Given their impact on intestinal homeostasis, a number of groups have studied ILC3s in human disease and have found them to be dysregulated in inflammatory bowel diseases such as ulcerative colitis and Crohn’s disease [63] [16]. While studies are generally in agreement with a pathological role for ILCs in mediating mouse models of colitis [64, 65], it is important to note that ascribing a direct role for ILC3s in the etiology of human intestinal diseases is difficult. As ILC3s are vastly outnumbered by adaptive immune cells that occupy the same anatomical niche and share largely identical effector functions, their influence on some facets of GI homeostasis may be dispensable. Indeed, while NKp46+ ILC3s are essential for defense against Citrobacter rodentium in T cell-deficient mice, they become redundant in fully immune-competent hosts [66, 67]. IL-22 that is produced constitutively in ILC3s from RAG−/− mice becomes only transient in mature wild-type mice [55]. There is also some evidence of ILC redundancy in humans [68]. On the other hand, ILC3s were found in one study to have an essential role in promoting CD4+ T cell tolerance to commensal bacteria through their ability to process and present antigen [69]. Thus, it is likely that ILC3s play both redundant and nonredundant roles in GI tissue homeostasis, and considering their impact in the context of other immune cell types within the gut is important in evaluating these cells in human health and disease.
ILCs in Primate Immunodeficiency Lentiviral Infections
Distinct pathological events occur to the GI tract throughout HIV disease [2, 3, 70, 71], all of which contribute to a site that is irreparably damaged and permeable to immunostimulatory commensal bacteria that are otherwise segregated to the intestinal lumen. Damage to the GI barrier is closely linked to depletion of IL-17-producing cells in the gut [72,73,74,75]. Given that ILC3s are recognized sources of this cytokine, there has been interest in characterizing these cells in HIV-infected subjects. To date, there have been only a handful of studies assessing these cells in treated or untreated subjects which are summarized in Table 1.
Two studies have examined ILC3s from gut tissue biopsies of viremic untreated HIV-1+ subjects. In the first, Zhang et al. employed immunohistochemistry to enumerate colonic IL-17-producing cells that were CD3− (presumably enriched for ILC3s) and CD3+. In both IL-17-producing populations, their numbers were reduced when compared to these cells from colonic biopsies of healthy control subjects [76]. The second study by Kloverpris et al. observed colonic Lineage−CD127+CD117+ ILC3 frequencies to be similar among HIV-1+ and healthy control subjects [77••]. In this study, however, it is relevant to note that treatment status for the particular HIV-1 cohort undergoing biopsies was not indicated.
Although ILCs are exceedingly rare in peripheral blood, the accessibility of this compartment allows for ILCs to be studied in cohorts where certain variables are more controlled. Studies of peripheral blood have revealed that ILC3s (Lineage−CD127+CD117+) are decreased in numbers in HIV-1+ untreated subjects, and importantly, correlate inversely with surrogate markers of GI epithelial damage [76, 77••]. The study performed by Kloverpris and colleagues was particularly comprehensive in that it was longitudinal and blood was able to be sampled at very early timepoints in HIV-1 disease course, 5–14 days after transmission. The overall dynamics emerging from this study indicated a strikingly rapid decline of ILC1, ILC2, and ILC3 subtypes in blood that coincided with peak viremia (~ 14 post-transmission) [77••]. In contrast to circulating CD4 T cell counts in these subjects, no rebound in blood ILC numbers was observed after viral set point [77••].
In a separate cohort, Kloverpris and colleagues also tracked the dynamics of blood ILCs after 2 years of virologically suppressive cART. When administered during chronic infection, cART allowed for partial reconstitution of circulating CD4 T cells and reduced inflammation, yet proportions of the three ILC populations in blood reconstituted incompletely [77••]. Only the ILC3 subset rebounded with cART to a significant degree, yet never to levels seen in healthy subjects [77••]. Interestingly, depletion of blood ILCs did not occur if cART was administered to subjects very early in disease course at 5–14 days post-transmission, indicating these cells could be preserved if peak viremia is abolished [77••].
While the above study described the dynamics of blood ILCs in response to long-term cART, how accurately these parallel biological processes in GI tract tissues is unclear. To date, three cross-sectional studies have assessed tissue-resident ILC3s in cART-treated subjects. Fernandes et al. observed numbers of CD3−IL-22+ cells in the colon to be similar among healthy control and cART-treated HIV-1+ subjects (some of which were on cART for 8 years) [78]. These findings are in agreement with a separate study that found no significant differences between numbers of colonic NKp44+ ILC3s in healthy control and cART-treated HIV-1+ subjects [79]. While these reports sampled only colonic tissue, Kramer and colleagues were able to assess ILC3s across the entire length of the digestive tract. Relative to these sites in healthy uninfected subjects, NKp44+ ILC3 frequencies in HIV-1+ cART-treated subjects were similar in the esophagus and stomach, increased in the duodenum, and reduced in the colon [46••].
Taken together, these studies suggest a general lack of consensus regarding ILC dynamics in settings of treated or untreated HIV-1 infection, particularly in tissues. It is thus relevant to ask what factors may contribute to this lack of consensus. For one, there are technical challenges inherent in assessing ILCs through intestinal pinch biopsies. Even at sites where ILCs are enriched, it is difficult to measure this rare cell population where low numbers of leukocytes are obtained. Secondly, there are methodological differences, both in how ILCs were defined and how they were quantified which potentially preclude the studies from being compared directly. For example, Kramer et al. measured c-Kit+ ILC3s proportionally in intestinal cell suspensions, whereas Fernandes et al. quantified CD3−IL-22+ cells in fixed tissue by immunohistochemistry [46••, 78]. To expand on this point, it is important to note that lymphocyte isolation from mucosal tissues is oftentimes incomplete, and proportional assessments may not be fully representative of true ILC numbers within the gut [80]. Thus, the field may benefit from histological assessments that enumerate ILCs directly in tissues to shed further light on this important issue.
Given the challenges noted above in human studies, the use of nonhuman primate models of AIDS has provided critical insights into perturbations that may occur to ILCs during immunodeficiency lentiviral infections. SIV infection of rhesus macaques recapitulates the most salient features of the HIV-1 disease course [81], and in all studies examining ILC3s in this setting, each point to an early and sustained depletion of these cells in the gut mucosa [22••, 82, 83, 84••, 85]. Both Reeves et al. and our group initially characterized ILC3s in chronically SIV-infected animals, with our group observing a correlation between loss of ILC3s in the gut and breaches to the gut mucosal barrier [82, 84••]. These were subsequently followed by two studies examining the particular dynamics at which the NKp44+ ILC3 subset was lost in the GI tract. Each of these found NKp44+ ILC3 frequencies to be significantly reduced as early as 6 days post-infection [85, 86], a striking observation given that these cells are refractory to HIV/SIV infection [84••]. In recent work, our group has observed ILC3s to be depleted at similar kinetics in SIV+ mesenteric lymph nodes (MLN). The impact of cART was also assessed in this study, where we observed frequencies of NKp44+ ILC3s to be similar in MLNs of healthy uninfected and SIV+ ART-treated animals. These findings contrast somewhat to those of Liyanage et al., in which NKp44+ ILC3s of SIV+ rhesus macaques receiving cART remained depleted in the rectum [86]. The underlying reasons for the discordance in these two studies are currently uncertain, but assessing ILC3s longitudinally in multiple anatomical sites before and after cART may shed light on this issue.
Nonhuman primate work has also guided current thought processes on how these cells may be functionally altered during immunodeficiency lentiviral infections. An observation linking many of these studies is that SIV infection appears to induce NKp44+ ILC3s to acquire cytotoxic functions [22••, 84••, 85, 86]. Although not to the degree of NKG2A+ NK cells, NKp44+ ILC3s in the SIV+ colon were observed to have greater capacity to express CD107a, perforin, and IFNγ upon stimulation [84••, 85].
Moreover, SIV infection appears to influence IL-17 and IL-22 production in ILC3s. We found in both the acute and chronic SIV+ MLN that even though the total numbers of ILC3s were decreased within mucosal tissues, IL-17 and IL-22 transcripts are increased in NKp44+ ILC3s directly ex vivo, and that the few remaining ILC3s express higher amounts of IL-17 and IL-22 protein upon stimulation [22••]. Human data also suggest elevated IL-17 and IL-22 production in stimulated colonic ILC3s from chronic HIV-1+ subjects [46••, 87]. While these findings are somewhat contrary to the recognized link between IL-17-producing cells and GI integrity, it is certainly clear that damage to the GI tract in HIV/SIV infections is associated with significant numerical and functional alterations to ILC populations in the gut mucosa.
Mechanisms of ILC Loss in HIV/SIV Infections
ILCs do not express HIV/SIV entry receptors and do not harbor viral DNA [84••]. Thus, a logical question emerging from the above studies is how these cells are depleted so rapidly without being directly targeted by the virus. For one, it is clear that ILC depletion in HIV/SIV infections is the result of cell death. We and others have observed ILCs at multiple anatomical sites to exhibit elevated levels of caspase-3 and robust gene signatures of apoptosis [22••, 49, 77••, 85]. These apoptotic signatures coincide with peak viremia, production of type I interferons, and expression of interferon-stimulated genes (ISGs). Indeed, we and others have found upregulation of multiple ISGs in ILC3s at 6–14 days post HIV/SIV infection [22••, 77••], timepoints at which plasma levels of IP-10 and IFNα are known to spike [88]. Importantly, CD3−IL-17+ cells in the gut are preserved in nonprogressively, SIV-infected sooty mangabeys, which do not exhibit chronic elevation of type I interferons or ISGs [82, 89].
While the potential mechanisms of IFNα-mediated ILC3 death are not entirely clear, Zheng et al. explored this question in humanized mice, which develop functional human ILC3s and recapitulate important aspects of disease course upon HIV-1 infection [49]. Similar to studies in HIV/SIV+ humans and rhesus macaques, they found HIV-associated depletion of ILC3s to coincide with caspase-3 upregulation [49]. ILC3 loss could be partially rescued through direct abolishment of plasmacytoid dendritic cell-derived IFNα, which induced CD95 (Fas) expression on ILC3s and sensitized them to apoptosis [49]. Subsequently, Kloverpris et al. have found CD95 to be upregulated on blood ILC3s in chronic HIV-1 infection [77••], suggesting IFNα may mediate ILC death via Fas/Fas ligand interactions.
It is important to point out that loss of ILCs is not a generalized feature of all viral infections, and thus mechanisms independent of IFNα may also contribute [32, 77••]. The persistent inflammatory environment in HIV/SIV infection uniquely alters many physiological processes in the GI tract, one of which is altered catabolism of tryptophan metabolites by the enzyme indoleamine 2,3-dioxygenase 1 (IDO1) [90, 91]. As tryptophan catabolites decrease TH17/TREG ratios [92], and factors that influence TH17 and ILC3s often overlap, Reeves et al. explored SIV-associated links to IDO1 activity and ILC3 function [84••]. Here, IDO1 transcripts were found to be increased in gut biopsies from SIV+ rhesus macaques, which correlated inversely with numbers of NKp44H ILC3s in the colon [84••]. Moreover, treatment with tryptophan catabolites in vitro suppressed IL-17 production in ILC3s in a dose-dependent manner [84••], highlighting an additional mechanism that may contribute to HIV/SIV-associated ILC3 cell death.
Surprisingly, we recently found that depletion of ILCs occurs even when salient features of HIV/SIV pathology are induced in the absence of SIV infection. In this study, we observed that experimental depletion of CD4 T cells in otherwise healthy rhesus macaques led to drastic depletion of ILC3s in the MLN [22••]. Profound ILC deficiencies were also observed in the blood of human subjects with idiopathic CD4 lymphocytopenia (ICL), a disease characterized by durable CD4 T cell deficiency in the absence of any infectious component [22••]. These findings reveal striking parallels to ILC loss in HIV/SIV infections, and also raise the question of potential mechanisms by which CD4 T cells could impact ILC homeostasis directly. A promising candidate may be IL-2, a cytokine produced by CD4 T cells which induces ILC proliferation [44, 93]. Interestingly, daclizumab, a CD25 blocker known to reduce multiple sclerosis-associated inflammation resulted in decreased numbers of blood ILCs (but not T cells) in MS subjects [94]. Future studies will be needed to uncover the precise cross-talk between ILCs and CD4 T cells, although taken together these studies suggest loss of ILCs may be due to a number of bystander effects induced by HIV/SIV replication.
Conclusions
A large body of evidence in humans and mice now show that ILCs, particularly the ILC3 subset, play a critical role in maintaining GI tract anatomy and physiology. It is thus not surprising that, in HIV/SIV infections, alterations to GI tract anatomy and physiology are associated with rapid decline and functional alterations to ILCs. The exact degree to which gut ILC3 loss contributes to HIV/SIV-associated intestinal pathology is unclear, given that IL-17-producing CD4, CD8, γδ, and MAIT cells are depleted in the GI tract at similar rates [72, 74, 95]. While functionally similar T cells may outnumber ILC3s in the gut, histological observations in human GI tissues have revealed CD3+ and CD3− cells actively producing IL-17 to be present at merely 2:1 ratios at steady state [49, 78]. Thus, a significant amount of IL-17 produced from the hematopoietic pool in humans could be ILC3-derived. It is, therefore, important to evaluate this population in the context of recent therapeutic modalities such as IL-7 and IL-21, or probiotic supplementations that aim to enhance GI tract physiology in cART-treated HIV-1+ subjects [72, 96, 97]. In additional work, enhanced efficacy of certain adjuvant formulations in combination with the ALVAC-SIV vaccine was associated with ILC3 expansion in the rectum [98].Thus, ILCs influence a number of biological processes with direct relevance to HIV pathogenesis and vaccine approaches, and future questions arising from these studies are rife for exploration.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200(6):749–59.
Rodgers VD, Fassett R, Kagnoff MF. Abnormalities in intestinal mucosal T cells in homosexual populations including those with the lymphadenopathy syndrome and acquired immunodeficiency syndrome. Gastroenterology. 1986;90(3):552–8.
Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434(7037):1148–52.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–71.
Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. 2017;23(11):1271–6.
Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest. 2011;121(3):998–1008.
Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. 2013;119:51–83.
Klatt NR, Canary LA, Sun X, Vinton CL, Funderburg NT, Morcock DR, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J Clin Invest. 2013;123(2):903–7.
Estes JD, Reilly C, Trubey CM, Fletcher CV, Cory TJ, Piatak M Jr, et al. Antifibrotic therapy in simian immunodeficiency virus infection preserves CD4+ T-cell populations and improves immune reconstitution with antiretroviral therapy. J Infect Dis. 2015;211(5):744–54.
Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457(7230):722–5.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70.
Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464(7293):1362–6.
Hazenberg MD, Spits H. Human innate lymphoid cells. Blood. 2014;124(5):700–9.
Martin CE, Spasova DS, Frimpong-Boateng K, Kim HO, Lee M, Kim KS, et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity. 2017;47(1):171–82 e4.
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14(3):221–9.
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350(6263):981–5.
Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211(3):563–77.
Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36(1):55–67.
Beziat V, Duffy D, Quoc SN, Le Garff-Tavernier M, Decocq J, Combadiere B, et al. CD56brightCD16+ NK cells: a functional intermediate stage of NK cell differentiation. J Immunol. 2011;186(12):6753–61.
•• Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. 2018;48(5):1060 This provides a multi-dimensional phenotypic analysis of ILCs across multiple human tissues. Likely the most comrehensive chacterization to date in humans.
•• Mudd JC, Busman-Sahay K, DiNapoli SR, Lai S, Sheik V, Lisco A, et al. Hallmarks of primate lentiviral immunodeficiency infection recapitulate loss of innate lymphoid cells. Nat Commun. 2018;9(1):3967 This study sheds important light on potential mechanisms of SIV/HIV-associated ILC depletion.
Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol. 2016;17(4):451–60.
Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, et al. CD4+ group 1 innate lymphoid cells (ILC) form a functionally distinct ILC subset that is increased in systemic sclerosis. J Immunol. 2016;196(5):2051–62.
Zhao J, Cheng L, Wang H, Yu H, Tu B, Fu Q, et al. Infection and depletion of CD4+ group-1 innate lymphoid cells by HIV-1 via type-I interferon pathway. PLoS Pathog. 2018;14(1):e1006819.
Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. 2017;168(6):1086–100 e10.
Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, et al. A progenitor cell expressing transcription factor RORgammat generates all human innate lymphoid cell subsets. Immunity. 2016;44(5):1140–50.
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16.
Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37(4):634–48.
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17(6):636–45.
Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. 2016;17(6):626–35.
Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. 2016;17(6):646–55.
Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213(4):569–83.
Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283–95.
Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519(7542):242–6.
Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. 2013;14(6):536–42.
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502(7470):245–8.
Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17(7):765–74.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174(5):1054–66.
Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10(1):75–82.
Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10(1):66–74.
Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, et al. RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity. 2013;38(6):1223–35.
Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A. 2010;107(24):10961–6.
Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. 2015;43(1):146–60.
•• Kramer B, Goeser F, Lutz P, Glassner A, Boesecke C, Schwarze-Zander C, et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017;13(5):e1006373 Likely the most comprehensive assessment of ILC3s in human intestinal tissues.
Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336(6086):1321–5.
Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118(2):534–44.
Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–9.
Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. 2000;118(6):1001–11.
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14(3):275–81.
Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29(6):958–70.
Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33(5):752–64.
Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211(8):1571–83.
Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. 2018;554(7691):255–9.
Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep. 2017;7(1):3980.
Perdew GH, Babbs CF. Production of Ah receptor ligands in rat fecal suspensions containing tryptophan or indole-3-carbinol. Nutr Cancer. 1991;16(3–4):209–18.
Zelante T, Iannitti RG, Fallarino F, Gargaro M, De Luca A, Moretti S, et al. Tryptophan feeding of the IDO1-AhR axis in host-microbial symbiosis. Front Immunol. 2014;5:640.
Hubbard TD, Murray IA, Bisson WH, Lahoti TS, Gowda K, Amin SG, et al. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci Rep. 2015;5:12689.
Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334(6062):1561–5.
Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2011;13(2):144–51.
Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36(1):92–104.
Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208(6):1127–33.
Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2014;7(1):143–54.
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464(7293):1371–5.
Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol. 2016;17(2):179–86.
Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med. 2015;212(11):1869–82.
Vely F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17(11):1291–9.
Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498(7452):113–7.
Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5(193):193ra91.
Mudd JC, Brenchley JM. Gut mucosal barrier dysfunction, microbial dysbiosis, and their role in HIV-1 disease progression. J Infect Dis. 2016;214(Suppl 2):S58–66.
Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112(7):2826–35.
Nilssen DE, Muller F, Oktedalen O, Froland SS, Fausa O, Halstensen TS, et al. Intraepithelial gamma/delta T cells in duodenal mucosa are related to the immune state and survival time in AIDS. J Virol. 1996;70(6):3545–50.
Cosgrove C, Ussher JE, Rauch A, Gartner K, Kurioka A, Huhn MH, et al. Early and nonreversible decrease of CD161++ /MAIT cells in HIV infection. Blood. 2013;121(6):951–61.
Schuetz A, Deleage C, Sereti I, Rerknimitr R, Phanuphak N, Phuang-Ngern Y, et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014;10(12):e1004543.
Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, et al. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J Clin Invest. 2015;125(9):3692–703.
•• Kloverpris HN, Kazer SW, Mjosberg J, Mabuka JM, Wellmann A, Ndhlovu Z, et al. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity. 2016;44(2):391–405 The most comprehensive characterization of blood ILCs in HIV-1 disease course.
Fernandes SM, Pires AR, Ferreira C, Foxall RB, Rino J, Santos C, et al. Enteric mucosa integrity in the presence of a preserved innate interleukin 22 compartment in HIV type 1-treated individuals. J Infect Dis. 2014;210(4):630–40.
Dillon SM, Castleman MJ, Frank DN, Austin GL, Gianella S, Cogswell AC, et al. Brief report: inflammatory colonic innate lymphoid cells are increased during untreated HIV-1 infection and associated with markers of gut dysbiosis and mucosal immune activation. J Acquir Immune Defic Syndr. 2017;76(4):431–7.
Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell. 2015;161(4):737–49.
Estes JD, Wong SW, Brenchley JM. Nonhuman primate models of human viral infections. Nat Rev Immunol. 2018;18(6):390–404.
Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 2012;5(6):646–57.
Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. 2012;5(6):658–69.
•• Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, et al. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood. 2011, 12, 3321;118:–30 The first characterization of the dynamics of colon NKp44+ ILC3s in SIV infection.
Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, et al. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog. 2014;10(12):e1004551.
Liyanage NP, Gordon SN, Doster MN, Pegu P, Vaccari M, Shukur N, et al. Antiretroviral therapy partly reverses the systemic and mucosal distribution of NK cell subsets that is altered by SIVmac(2)(5)(1) infection of macaques. Virology. 2014;450–451:359–68.
Kim CJ, Nazli A, Rojas OL, Chege D, Alidina Z, Huibner S, et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol. 2012;5(6):670–80.
Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83(8):3719–33.
Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest. 2009;119(12):3556–72.
Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2(32):32ra6.
Fuchs D, Moller AA, Reibnegger G, Werner ER, Werner-Felmayer G, Dierich MP, et al. Increased endogenous interferon-gamma and neopterin correlate with increased degradation of tryptophan in human immunodeficiency virus type 1 infection. Immunol Lett. 1991;28(3):207–11.
Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183(4):2475–83.
Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. 2011;17(5):604–9.
Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med. 2012;4(145):145ra06.
Gaardbo JC, Hartling HJ, Thorsteinsson K, Ullum H, Nielsen SD. CD3+CD8+CD161high Tc17 cells are depleted in HIV-infection. AIDS. 2013;27(4):659–62.
Thiebaut R, Jarne A, Routy JP, Sereti I, Fischl M, Ive P, et al. Repeated cycles of recombinant human interleukin 7 in HIV-infected patients with low CD4 T-cell reconstitution on antiretroviral therapy: results of 2 phase II multicenter studies. Clin Infect Dis. 2016;62(9):1178–85.
Micci L, Ryan ES, Fromentin R, Bosinger SE, Harper JL, He T, et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Invest. 2015;125(12):4497–513.
Vaccari M, Gordon SN, Fourati S, Schifanella L, Liyanage NP, Cameron M, et al. Adjuvant-dependent innate and adaptive immune signatures of risk of SIVmac251 acquisition. Nat Med. 2016;22(7):762–70.
Funding
Funding for this study was provided in part by the Division of Intramural Research/NIAID/NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
The content of this publication does not necessarily reflect the views or policies of DHHS, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on HIV Pathogenesis and Treatment
Rights and permissions
About this article

Cite this article
Mudd, J.C., Brenchley, J.M. Innate Lymphoid Cells: Their Contributions to Gastrointestinal Tissue Homeostasis and HIV/SIV Disease Pathology. Curr HIV/AIDS Rep 16, 181–190 (2019). https://doi.org/10.1007/s11904-019-00439-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-019-00439-4