
Abstract
Purpose of Review
Recent efforts to characterize hematologic cancers with genetic and molecular detail have largely relied on mutational profiling via next-generation sequencing (NGS). The application of NGS-guided disease prognostication and clinical decision making requires a basic understanding of sequencing advantages, pitfalls, and areas where clinical care might be enhanced by the knowledge generated. This article identifies avenues within the landscape of adult acute lymphoblastic leukemia (ALL) where mutational data hold the opportunity to enhance understanding of disease biology and patient care.
Recent Findings
NGS-based assessment of measurable residual disease (MRD) after ALL treatment allows for a sensitive and specific molecular survey that is at least comparable, if not superior, to existing techniques. Mutational assessment by NGS has unraveled complex signaling networks that drive pathogenesis of T-cell ALL. Sequencing of patients with familial clustering of ALL has also identified novel germline mutations whose inheritance predisposes to disease development in successive generations.
Summary
While NGS-based assessment of hematopoietic malignancies often provides actionable information to clinicians, patients with acute lymphoblastic leukemia are left underserved due to a lack of disease classification and prognostication schema that integrate molecular data. Ongoing research is positioned to enrich the molecular toolbox available to clinicians caring for adult ALL patients and deliver new insights to guide therapeutic selection, monitor clinical response, and detect relapse.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With more than 6000 new diagnoses expected in the USA each year, acute lymphoblastic leukemia (ALL) is the second most common acute leukemia of adults (https://seer.cancer.gov/statfacts/html/alyl.html). The disease is characterized by the proliferation of lymphoid precursor cells and propagated by the acquisition of chromosomal alterations and driver mutations in critical genes. ALL is the most common malignancy of childhood, where cure rates currently exceed 90% with multi-agent chemotherapy [1]. By contrast, the success of chemotherapy in adult ALL is less dramatic, partly due to intrinsically more adverse risk disease in this population, but also due to patient-specific factors such as older age and comorbidity which limit the capacity of older ALL patients to tolerate multi-agent chemotherapy. Adult-specific multi-agent chemotherapeutic regimens have historically produced cure rates between 40 and 50% despite high rates of initial complete remission, although the more routine adoption of truly pediatric regimens for the younger adult population (those < 40 years) appears to have resulted in a marked improvement in outcome [2, 3]. Despite some successes, relapse remains a significant problem and strategies aimed to detect, characterize, and reduce the incidence of relapse are an area of active investigation.
ALL manifests without pathogonomonic clinical features and patient presentations are often characterized by nonspecific symptoms including fevers, fatigue, diaphoresis, bone pain, or bleeding complications. The diagnostic workup entails a thorough history and physical exam, laboratory studies, imaging to survey for extramedullary involvement, lumbar puncture with analysis of the cerebrospinal fluid, and bone marrow aspiration and biopsy. Disease subtype classification and risk stratification is accomplished by morphologic examination, cytogenetic analysis, and flow cytometry immunophenotyping [4]. The 2016 revision to the World Health Organization classification schema further defined two new categories for T-lymphoblastic (T-ALL) and B-lymphoblastic ALL (B-ALL), and describes provisional entities within the B-ALL categorization including B-ALL with intrachromosomal amplification of chromosome 21 and B-ALL with a gene expression profile resembling BCR-ABL + ALL (Ph-like ALL) [5].
Molecular profiling for patients with ALL remains an area of active research. When compared to solid tumors, hematologic malignancies are relatively bland with respect to the number and diversity of mutational events [6]. In recent years, the therapeutic toolbox for solid tumors has morphed from one heavily reliant on combination chemotherapy regimens to one rooted in the use of targeted therapies directed at oncogenic driver mutations. The pace of targeted therapy development for hematologic malignancies has generally lagged behind, and, until recently, disproportionally favored chronic myeloid and lymphoid leukemias for which imatinib and ibrutinib (among others) have significantly improved outcomes. Since 2017, several new treatments have also been FDA-approved for the treatment of acute myeloid leukemia (AML). Use of many of these therapies relies on the detection of mutations in specific targetable genes for which novel therapies have been designed. Based on guidelines from the European Leukemia Net and the National Comprehensive Cancer Network, AML patients should undergo comprehensive molecular sequencing of genes with immediate implications for targeted therapeutics, including c-KIT, FLT3-ITD, FLT3-TKD, NPM1, CEBPA, IDH1/2, RUNX1, ASXL1, and TP53 [7, 8]. While gene-by-gene, PCR-based strategies are feasible, testing with larger panels using next-generation sequencing (NGS) is well-validated and could potentially identify additional informative mutations [9]. For these reasons, the European Leukemia Net 2017 update supports panel-based NGS for molecular prognostication and target identification in patients with myeloid leukemia.
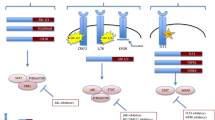
According to the National Comprehensive Cancer Network guidelines for both adult and pediatric ALL, upfront NGS-based mutational workup is not uniformly recommended [10, 11]. This is likely due to the heterogeneity that exists among sequencing methodologies, a lack of harmonization regarding a “minimum” set of target genes to be included in any given assay, and center-dependent factors such as operator expertise and laboratory limitations. However, de facto practices at many major academic centers have led to the collection of mutational data and guided strategies for the provision of targeted therapies in either compassionate use settings or through formal clinical protocols. A model for how acquisition of molecular data has informed treatment selection for Ph-like ALL is depicted in Fig. 1. Several articles in the literature have also comprehensively summarized classification schema, prognosticating factors, available treatments, and considerations surrounding allogeneic stem cell transplantation in patients with ALL [12,13,14]. In this a review, we consider emerging uses for NGS-based mutational profiling in patients with acute lymphoblastic leukemia, with a focus on adult ALL mutational profiling. We will highlight three exciting avenues within the ALL landscape that have been delineated by NGS approaches and that hold implications for disease prognostication and clinical decision making: (1) molecular measurable residual disease testing, (2) disentangling the complicated biology of T-ALL, and (3) ALL germline predisposition syndromes.

Targeted kinase inhibition in Philadelphia chromosome (Ph)-like acute lymphoblastic leukemia. Common signaling pathways in Ph-like ALL can be categorized by (1) activating lesions in JAK-STAT pathways, including CRLF2, IL7R, EPOR rearrangements, JAK mutations, and SH2B3 deletion/mutation (not shown); (2) oncogenic fusion proteins involving ABL-class tyrosine kinases, and (3) other rearrangements involving kinases such as FLT3, FGFR1, and NTRK3 which propogate survival and proliferation of malignant cells via STAT-mediated Ras/PI3K predominant signaling. A rational approach to targeted kinase inhibition is shown based on a review of the literature [71,72,73,74,75,76,77]. Of note, Ras pathway mutations are not specific to Ph-like ALL and are present in other ALL subtypes. For simplicity and emphasis on therapeutic targets, kinase cascades in the pathways shown have been condensed and certain intermediate proteins omitted. Image created with BioRender
Molecular MRD in ALL
Leukemic relapse stems from the persistence of malignant cells after completion of treatment that are capable of propagating the return of clinically overt disease. Measurable residual disease (MRD) is the term used to describe the detection of small populations of clinically relevant leukemia phenotypes capable of resulting in leukemic relapse. Detection of MRD has been shown to have powerful prognostic implications in a variety of clinical contexts. Techniques to characterize and quantitate MRD in ALL were first widely validated and used in pediatric ALL cohorts, and eventually, more data in the adult population has allowed MRD to gain broad adoption across the spectrum of age. MRD testing can be achieved using a variety of techniques including multiparameter flow cytometry (MFC), real-time quantitative polymerase chain reaction (PCR), as well as NGS. Regardless of the modality used (discussed in detail below), large meta-analyses with cumulative totals including thousands of individual ALL patients have demonstrated markedly superior outcomes in terms of event-free and overall survival for those who attain states of MRD negativity (MRD −) compared to individuals who harbor detectable disease (MRD +) at the end of induction or consolidation [15]. The most straightforward approach for measuring MRD utilizes highly sensitive real-time quantitative PCR techniques with primers that probe for regions of the rearranged immunoglobulin and T-cell receptor genes in cells that comprise the ALL clone. MFC uses lasers to detect optically active antibodies attached to cell surfaces and define a leukemia-associated immunophenotype (LAIP). An LAIP can be identified at the time of initial diagnosis and can therefore allow definition of an MRD population for a majority of both B-ALL and T-ALL patients. The sensitivity of MFC varies, with early 4–6 multicolor modalities possessing a limit of detection of 1 abnormal cell in a background of 103 or 104 normal cells. The newer 8–10 multicolor flow cytometry techniques can provide improved sensitivity on the order of 1 abnormal cell in a background of 105 normal cells [16, 17]. The major advantage to MFC-based MRD assessment over other techniques is its wide application; about 90% of patients can be assigned a LAIP at the time of first presentation. However, a critical limitation of MFC-based MRD is the impact of therapy-associated changes in clonal composition, which can result in phenotypic shifts in both non-leukemic cells and in the LAIP-associated MRD populations. Such changes can lead to both false positive and false negative reports for MRD status at the time of repeat evaluation and create a considerable clinical conundrum for patients and caregivers upon repeat survey [18].
As use of NGS-guided molecular assessment of both benign and malignant hematologic disorders continues to win favor [19], the optimization and clinical integration of NGS-driven MRD in ALL is underway [20]. Massively parallel, clinical NGS can be employed through numerous iterations, including targeted exon or “hotspot” sequencing, whole genome or whole exon sequencing, and transcriptome-focused methods via RNA sequencing. The sensitivity of NGS-based MRD assessments can range from 1 in 105 or 106 depending on the amount of input genetic material and the bioinformatic analysis technique employed. As with other NGS applications, NGS-based MRD is attractive due to its scalability and high throughput via automated wet lab procedures reducing human “hands-on” time [21]. Additionally, with new “barcoding” techniques using unique molecular indices to watermark individual nucleic acid strands, samples from multiple patients or clinical timepoints can be pooled into single sequencing reaction without worry of contamination [22].
In the real world, targeted sequencing approaches often take the form of commercially available panels and includes genes that, when mutated, offer actionable prognostic or therapeutic information. These modalities, employed upfront at diagnosis, are now commonplace. The design and analysis of NGS MRD assays are drastically different depending on whether they are intended for myeloid or lymphoid malignancies. AML, for example, is a mutationally heterogeneous, polyclonal disease whose clonal architecture changes in response to therapy [23]. As a result, NGS-based AML MRD panels need to cover a large collection of relevant target genes in order to assign a mutational profile in each patient to be tracked longitudinally over time [24]. Genes encoding cell surface proteins that serve as therapy targets are generally poor MRD markers. For example, molecular analysis of patients enrolled in the landmark RATIFY trial establishing midostaurin as an effective treatment for FLT3 mutant AML found that 46% of patients relapsed with FLT3 negative clones, emphasizing the lack of utility for FLT3 mutational events in the context of MRD monitoring [25].
By contrast, NGS-guided MRD in the context of lymphoid malignancies is more straightforward. Because ALL demonstrates a lower degree of clonal heterogeneity, sequencing libraries can be easily constructed by targeting regions flanking the immunoglobulin (Ig) or T-cell receptor loci (TCR), functionally providing a digital readout of traditional PCR techniques. Junctional rearrangement sequences (V-D-J domains) can be detected in ~ 95% of ALL clones from patients, and this relative uniformity of sequence reads makes MRD detection possible for a majority of patients with ALL [26]. Because of the monoclonal nature of malignant lymphoid populations, a collaborative involving several European research groups with expertise in ALL MRD assay design, known as The EuroClonality-NGS Consortium (www.euroclonalityngs.org), formed in order to validate and standardize NGS assays in ALL. Their methodological guidelines for NGS-based ALL MRD were published in 2019, providing a harmonized benchmark for future research in the field. [27] Accordingly, the US FDA has authorized use of a commercial product known as the clonoSEQ Assay (Adaptive Biotechnologies, Seattle WA), a multiplexed PCR and NGS assay to identify and quantify MRD in B-cell malignancies (https://www.ajmc.com/view/fda-clears-cloneseq-to-detect-mrd-in-chronic-lymphocytic-leukemia). More recently, reports comparing the properties and utility of NGS vs. flow cytometry-based MRD in cohorts of ALL patients have favored strategies based upon NGS. The pediatric ALL literature is replete with examples of NGS-augmented MRD detection and clinical decision making, including robust data for patients with both B-ALL and T-ALL [28, 29]. Table 1 provides an overview on the relatively small body of literature using NGS-based MRD assays in the adult ALL patient population.
The concept of MRD is primarily one that centers around practical “actionability.” Namely, is MRD prophecy, or does it represent a modifiable risk factor? Large meta-analyses in both adult and pediatric populations have underscored the importance of achieving MRD negativity in ALL [30, 31]. An early trial from Germany demonstrated the potential to employ blinatumomab, a bispecific T cell-engager (BiTE) antibody that recognizes CD19 on one side and CD3 on the other, facilitating enhanced T- cell recognition of malignant B-lymphoid residual disease, as a salvage therapy to eliminate residual disease after induction or consolidation. Of 21 patients treated, 16 became MRD − , yielding a response rate of 80%. All MRD responses occurred by the end of the first cycle of blinatumomab therapy. Follow-up at 33 months revealed that 61% of MRD responders avoided disease recurrence. While blinatumomab first gained FDA approval for use in Philadelphia-chromosome negative (Ph −) relapsed/refractory B-ALL in 2014 based on the results depicting a 43% response rate [32], follow-up trials in Philadelphia-chromosome positive (Ph +) and pediatric cohorts enabled the expansion of FDA approval to a wider patient population [33, 34]. Subsequently, a pivotal trial from the German-Austrian group using blinatumomab to facilitate clearance of MRD + disease studied 116 patients with B-ALL who had achieved CR but who remained MRD + when assessed by PCR or flow cytometry. After treatment with blinatumomab, 78% of patients became MRD − . Those who became MRD − with treatment had a relapse-free survival of 23.6 months compared to 5.7 months for those who remained MRD + , with respective overall survivals of 38.9 months and 12.5 months [35]. This led to FDA approval of blinatumomab in early 2018 for ALL with MRD + and notably was the first drug to gain approval on the basis of MRD status [36].
An additional antibody-based therapy known as inotuzumab ozogamicin, which targets CD22 on B-cells, has also yielded promising results both for patients with relapsed/refractory ALL, and specifically for patients with MRD + . The antibody–drug conjugate was approved after a 2017 phase III trial in adults with either Ph + or Ph − relapsed/refractory ALL achieved a CR rate of 80% vs. 29.4% who achieved CR in an identically sized control arm who received standard intensive chemotherapy [37]. As with blinatumomab, patients receiving inotuzumab ozogamicin also achieved significantly higher rates of MRD negativity (78%) than the control group 28%.
From the trials mentioned above, a logical question might arise about the utility of stem cell transplantation in the setting of MRD status for patients who received either blinatumomab or inotuzumab ozogamicin. As noted by Kantarjian and colleagues, inotuzumab ozogamicin enabled 41% of patients to proceed to transplant while only 11% of patients in the standard of care arm received an allograft [37]. Thus, inotuzumab ozogamicin may reasonably be utilized as a bridge to transplant strategy. Also, roughly half of the patients in pivotal blinatumomab trials mentioned above proceeded to transplant [38]. While MRD status appears to be among the most important prognostic factors in determining overall survival in ALL [31, 39, 40], provocative questions arise regarding the potential for agents like blinatumomab or inotuzumab ozogamicin to spare patients the morbidity and mortality of transplant if MRD − status is achieved. Although controversial, it is generally accepted that patients with detectable MRD would derive benefit from transplantation [36, 41]. More recently, the CALBG 10,403 study, which in short, demonstrated the benefit of treating adult ALL with pediatric-inspired regimens, also revealed that patients who achieve MRD − after induction performed well without an allogeneic transplant [3]. What is less clear is whether patients who might convert from MRD + to MRD − states with agents such as blinatumomab or inotuzumab ozogamicin could safely forego transplantation and enjoy similar rates of relapse-free and overall survival. An ongoing Alliance Study A41501 (https://clinicaltrials.gov/ct2/show/NCT03150693) seeks to evaluate the addition of novel agents such as inotuzumab ozogamicin to the CALGB 10,403 chemotherapeutic backbone. If a higher proportion of patients are able to attain MRD − , perhaps more individuals could avoid the rigors of transplant but derive similar survival benefits.
Unraveling Consequential Pathways in T-ALL
T-ALL comprises about 25% of cases in adults and is generally associated with poorer outcomes, likely due to the inherent biology of the disease and a smaller selection of novel therapies directed against T-cells as opposed B-cells [12]. For example, popular agents in the MRD + setting, such as blinatumomab, work by promoting T-cell directed killing of B-cells, and will obviously not be effective when the target cell population is not comprised of B lymphoblasts [36]. Recently, additional consideration has been given to defining oncogenic genotypes and delineating their effects on the pathogenesis of T-ALL. Recognized T-ALL oncogenic pathways include constitutive activation of the NOTCH1 circuit by a variety of NOTCH1/FBXW7 mutations, as well as pro-proliferative RAS/RAF/MEK and PTEN/Akt/mTOR mutations. Detection of NOTCH1/FBXW7 mutations in the absence of RAS or PTEN variants predicts a favorable outcome and is found in almost 50% of adult T-ALL [42]. Conversely, the absence of NOTCH1/FBXW7 or the presence of mutations in the RAS/PTEN pathway is associated with a poorer prognosis. In combination with MRD-based assessment, this additional layer of independent prognostic data may help to guide clinicians in clinical decision making, such as whether stem cell transplant should be pursued in first complete remission [30, 43].
Recognition of specific disease subsets, such as the early thymic precursor (ETP) ALL molecular subgroup, is another example of the way in which NGS characterization can enhance understanding of disease biology and outcomes. ETP ALL is a subset of T-ALL originally discovered in pediatric patients but subsequently also described in adults with an analogous transcriptional landscape. It has a characteristic immunophenotype demonstrating absent/nearly absent T-lymphocyte markers and positivity for at least one hematopoietic stem cell or myeloid antigen. Early sequencing efforts in childhood ETP ALL uncovered a mutational landscape that is distinct from non-ETP disease. Notably, pediatric ETP ALL was found to be enriched for activating mutations in genes regulating RAS and cytokine receptor signaling and inactivating mutations in genes related to hematopoietic development and epigenetic modification of histones [44]. Efforts aimed at describing the genomic composition of adult ETP ALL have yielded similar patterns, and mutations in genes traditionally associated with myeloid malignancies, such as DNTM3A and RUNX1, were particularly remarkable [45, 46].
While children with ETP ALL were initially thought to have poor outcomes secondary to chemotherapy resistance [47], further analyses have concluded that the high-risk biological features could be neutralized by timely MRD-directed salvage therapies [48]. An influential 2017 study by Bond and colleagues examined similar factors in a large adult cohort of T-ALL patients (n = 213), 47 of whom had ETP ALL. As seen in the pediatric patient population, adults with the ETP phenotype had markedly higher resistance to corticosteroid and chemotherapy regimens, and tended to demonstrate higher rates of bone marrow MRD positivity; excitingly, these patients demonstrated similar overall survival rates compared with non-ETP patients in the context of MRD guided treatment strategies (5-year survival of 59.6% for ETP, 66.5% for non-ETP, P = 0.33) [49]. Based on multivariate analysis, the authors concluded that despite a more resistant disease biology, the use of NGS-guided risk stratification, with prompt therapeutic intensification, and the decision to proceed to allograft in first complete remission was able to markedly enhance survival in the ETP group.
One final example regarding the way in which NGS may enhance our understanding of disease biology pertains to the presents of PHF6 in T-ALL. PHF6 is an X-linked tumor suppressor gene known to be part of the mutational fingerprint of T-ALL. PHF6 mutations have been shown to occur in 16% of pediatric and 38% of adult primary T-ALL samples [50]. Notably, such mutations are also present in 25% of ETP-ALL cases. If inactivated by such a mutation, PHF6 has been shown to enhance hematopoietic stem cell self-renewal and to propagate transcriptional networks associated with leukemic cell stem-cell activation. This facilitates hematopoietic stem cell reactivation/proliferation for prolonged periods after chemotherapy [51]. Additionally, some cases of PHF6-mutant T-ALL may share a common cell of origin with seemingly unrelated secondary malignancies, such as histiocytic neoplasms. We have previously described NGS-guided therapy selection in a patient with a rare histiocytic sarcoma which presented after stem cell transplant for T-ALL; this malignancy harbored a mutational profile suggesting a shared neoplastic cell of origin. Using sequencing to identify mutational composition and tumor mutational burden allowed for a deeper understanding of disease modeling and hypotheses surrounding the PHF6-mutant driven divergence of the malignant T-cell clone from a bipotential progenitor cell [52].
Germline Predisposition to ALL
While thorough efforts have been focused on the somatic mutational landscape of ALL, germline variants predisposing to the development of ALL and other hematopoietic malignancies have gained increased recognition in recent years. For decades, blood cancers have been noted to cluster in families, but only recently have the molecular drivers of hereditary hematopoietic malignancies (HHM(s)) begun to be elucidated [53, 54]. Although the incidence of pathogenic or likely pathogenic germline variants in pediatric oncology cohorts is about 8.5% [55], it is a misconception that cancer predisposition syndromes only manifest in childhood. For example, germline variants in DDX41, which likely represent the most common HHM and account for at least 1% of all myeloid malignancies, have been described almost exclusively in adulthood [56, 57]. At this time, the impact of HHM-associated inheritance and phenotypic manifestations is better studied for myeloid than lymphoid malignancies, and a provisional entry for “Myeloid Neoplasms with Germline Predisposition” was included in the 2016 World Health Organization classification revision [5]. However, the contribution of heritable variants and the future development of lymphoid disorders remains an area of active investigations. Table 2 highlights several important HHMs associated with predisposition to ALL and two high yield syndromes are discussed below.
The PAX5 gene codes for a DNA-transcription factor with a critical role in B-cell differentiation and somatically acquired variants, in the form of pathogenic single nucleotide mutations, copy number alterations, or chromosomal rearrangements are common. Disruption in native PAX5 function has been shown to arrest B-cell development [58, 59]. The 2013 report of two unrelated families with autosomal dominantly inherited pre-B ALL established PAX5 as a new HHM syndrome. In both kindreds, germline PAX5 variants segregated with chromosome 9p loss, leading to a hemizygous variant allele [60]. More recently, Duployez and colleagues described a novel R38H PAX5 germline variant in a family devastated by the development of B-cell precursor ALL in three children, ages 11, 17, and 25 and a post-transplant donor-cell derived relapse involving two of the siblings [61]. From reports thus far, it does not appear that HHMs driven by germline PAX5 variants have an antecedent hematologic phenotype, such as cytopenias, before the development of B-ALL. Parental carriers in the affected families were asymptomatic, and unlike other HHMs, immunodeficiency was not a feature of either the carrier or diseased state. Therefore, it is reasonable to postulate that the frequency of cases related to germline mutant PAX-5 driven ALL might be an underestimate, and the syndrome should be considered prior matched related donor workup for B-ALL. However, given the lack of clinical features before the onset of ALL and the recognition that heterogeneity exists within commercially available tests for familial hematopoietic malignancies [62], diagnosis remains challenging.
TP53 is among the most commonly mutated genes in human cancers, and pathogenic germline TP53 variants are diagnostic of the Li-Fraumeni syndrome, which results in an enhanced risk for development of multiple tumor types, most prominently sarcomas, but also adrenocortical carcinomas, breast cancer, central nervous system cancers, and hematopoietic malignancies [63, 64]. Within the hematologic landscape, germline TP53 variants are most prominently associated with low hypodiploid ALL. In a series of 140 hypodiploid ALL patients, TP53 mutations were highly prevalent and in one pediatric cohort, 43.3% of variants were of germline origin [65]. Hypodiploid ALL is generally subclassified by patterns of chromosomal loss, with near haploid disease harboring 24–31 chromosome and low hypodiploid harboring 32–39. Additionally, each of these two subtypes are enriched for two distinct mutational patterns, with RAS pathway mutations being common in near-haploid ALL and mutations in IKZF2 and IKZF3 in low hypodiploid ALL [66, 67]. Large surveys of pediatric ALL have demonstrated germline TP53 variants in pediatric hypodiploid ALL (estimated to contribute to about 2% of all cases in a series of 3801 children) [68], but previously unrecognized germline variants can also be found in adult oncology patients. One study of breast cancer survivors who developed therapy-related leukemias found that 21% of carried deleterious germline variants. Of the 47 subjects diagnosed with therapy-related leukemia with specimens available for analysis, BRCA1/2 lesions were the most common (10% of cases), while TP53 variants comprised an additional 6% of cases. Because many of the genes noted to be mutated in therapy-related leukemias function to maintain the DNA damage response, future work will likely reframe our understanding of such events to a model whereby individuals harboring germline variants in critical genes are particularly vulnerable to the genotoxic stress imparted by chemotherapy or radiation for primary tumors [69, 70].
Conclusions
With the more widespread adoption of molecular profiling for patients with all types of cancer, individuals with ALL are likely to continue to derive benefit from improved appreciation for the molecular drivers of disease, moreover, widespread adoption of uniform and sensitive techniques for assessment of MRD are likely to improve the choice of treatment for patients and drive escalation or de-escalation of therapeutic intensity in an increasingly data driven manner. Information derived from NGS mutational assessment both at the time of initial diagnosis, as well as at critical follow-up time points has already transformed the myeloid malignancy space and is poised to augment diagnosis, risk stratification, and relapse prevention in the future. Currently, the use of NGS in ALL most commonly takes the form of targeted panels that are employed upfront at academic centers for patients presenting with hematologic malignancies. We anticipate that further study of NGS data will help refine and clarify molecular subsets of ALL to augment traditional disease classification and risk assessment driven by cytogenetics and clinical features. NGS-based MRD for lymphoid malignancies has already been broadly adopted in the context of pediatric ALL, and also holds promise to enhance or replace flow cytometry practices as it requires less operational cost and expertise and is able to automated for high throughput workflow. Blinatumomab is currently approved as an MRD-directed ALL therapy, but detection of residual disease at this time is generally achieved by single gene PCR or flow-cytometry based methods. In the future, NGS-driven MRD assessment may integrate sequence detection for the immunoglobulin or T-cell receptor of the malignant clone with detection of additional mutational signatures to enhance molecular risk stratification. Lastly, we anticipate significant future opportunities for NGS to help widen the spectrum of genes involved in hereditary hematopoietic malignancies and the molecular mechanisms underlying their pathogenesis.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
References
Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–41.
Sive JI, Buck G, Fielding A, Lazarus HM, Litzow MR, Luger S, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br J Haematol. 2012;157(4):463–71.
Stock W, Luger SM, Advani AS, Yin J, Harvey RC, Mullighan CG, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood. 2019;133(14):1548–59.
Paul S, Kantarjian H, Jabbour EJ. Adult acute lymphoblastic leukemia. Mayo Clin Proc. 2016;91(11):1645–66.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Tallman MS, Wang ES, Altman JK, Appelbaum FR, Bhatt VR, Bixby D, et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17(6):721–49.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Alonso CM, Llop M, Sargas C, Pedrola L, Panadero J, Hervás D, et al. Clinical utility of a next-generation sequencing panel for acute myeloid leukemia diagnostics. J Mol Diagn. 2019;21(2):228–40.
Brown P, Inaba H, Annesley C, Beck J, Colace S, Dallas M, et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(1):81–112.
Brown PA, Wieduwilt M, Logan A, DeAngelo DJ, Wang ES, Fathi A, et al. Guidelines insights: acute lymphoblastic leukemia, Version 1.2019. J Natl Compr Canc Netw. 2019;17(5):414–23.
Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6):e577.
Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146–62.
DeAngelo DJ, Jabbour E, Advani A. Recent Advances in Managing Acute Lymphoblastic Leukemia. Am Soc Clin Oncol Educ Book. 2020;40:330–42.
Berry DA, Zhou S, Higley H, Mukundan L, Fu S, Reaman GH, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: a meta-analysis. JAMA Oncol. 2017;3(7):e170580.
Denys B, van der Sluijs-Gelling AJ, Homburg C, van der Schoot CE, de Haas V, Philippé J, et al. Improved flow cytometric detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia. 2013;27(3):635–41.
Theunissen P, Mejstrikova E, Sedek L, van der Sluijs-Gelling AJ, Gaipa G, Bartels M, et al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood. 2017;129(3):347–57.
Bartram J, Patel B, Fielding AK. Monitoring MRD in ALL: Methodologies, technical aspects and optimal time points for measurement. Semin Hematol. 2020;57(3):142–8.
Haferlach T. The time has come for next-generation sequencing in routine diagnostic workup in hematology. Haematologica. 2021;106(3):659–61.
Sherali N, Hamadneh T, Aftab S, Alfonso M, Tsouklidis N. Integration of next-generation sequencing in diagnosing and minimal residual disease detection in patients with Philadelphia chromosome-like acute lymphoblastic leukemia. Cureus. 2020;12(9):e10696.
Kruse A, Abdel-Azim N, Kim HN, Ruan Y, Phan V, Ogana H, et al. Minimal residual disease detection in acute lymphoblastic leukemia. Int J Mol Sci. 2020;21(3).
Roloff GW, Lai C, Hourigan CS, Dillon LW. Technical advances in the measurement of residual disease in acute myeloid leukemia. J Clin Med. 2017;6(9).
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78.
Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med. 2018;378(13):1189–99.
Schmalbrock LK, Dolnik A, Cocciardi S, Sträng E, Theis F, Jahn N, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021.
van der Velden VH, van Dongen JJ. MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. Methods Mol Biol. 2009;538:115–50.
Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33(9):2241–53.
Wu D, Sherwood A, Fromm JR, Winter SS, Dunsmore KP, Loh ML, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4(134):134ra63.
Kotrova M, van der Velden VHJ, van Dongen JJM, Formankova R, Sedlacek P, Brüggemann M, et al. Next-generation sequencing indicates false-positive MRD results and better predicts prognosis after SCT in patients with childhood ALL. Bone Marrow Transplant. 2017;52(7):962–8.
Beldjord K, Chevret S, Asnafi V, Huguet F, Boulland ML, Leguay T, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739–49.
Ravandi F, Jorgensen JL, O’Brien SM, Jabbour E, Thomas DA, Borthakur G, et al. Minimal residual disease assessed by multi-parameter flow cytometry is highly prognostic in adult patients with acute lymphoblastic leukaemia. Br J Haematol. 2016;172(3):392–400.
Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66.
Martinelli G, Boissel N, Chevallier P, Ottmann O, Gökbuget N, Topp MS, et al. Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol. 2017;35(16):1795–802.
von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381–9.
Gökbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522–31.
Curran E, Stock W. Taking a “BiTE out of ALL”: blinatumomab approval for MRD-positive ALL. Blood. 2019;133(16):1715–9.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740–53.
Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–8.
Bassan R, Spinelli O, Oldani E, Intermesoli T, Tosi M, Peruta B, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–62.
Bassan R, Brüggemann M, Radcliffe HS, Hartfield E, Kreuzbauer G, Wetten S. A systematic literature review and meta-analysis of minimal residual disease as a prognostic indicator in adult B-cell acute lymphoblastic leukemia. Haematologica. 2019;104(10):2028–39.
Dhédin N, Huynh A, Maury S, Tabrizi R, Beldjord K, Asnafi V, et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood. 2015;125(16):2486–96; quiz 586.
Petit A, Trinquand A, Chevret S, Ballerini P, Cayuela JM, Grardel N, et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood. 2018;131(3):289–300.
Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, Lambert J, Beldjord K, Lengliné E, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol. 2013;31(34):4333–42.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63.
Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer. 2013;52(4):410–22.
Neumann M, Greif PA, Baldus CD. Mutational landscape of adult ETP-ALL. Oncotarget. 2013;4(7):954–5.
Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–56.
Wood B, Winter S, KP D, M D, S C, B A, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) study AALL0434. Blood: 124; 2014. p. 1.
Bond J, Graux C, Lhermitte L, Lara D, Cluzeau T, Leguay T, et al. Early response-based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: a group for research on adult acute lymphoblastic leukemia study. J Clin Oncol. 2017;35(23):2683–91.
Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(4):338–42.
Wendorff AA, Quinn SA, Rashkovan M, Madubata CJ, Ambesi-Impiombato A, Litzow MR, et al. Loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov. 2019;9(3):436–51.
Roloff GW, Baron JI, Neppalli VT, Sait S, Griffiths EA. Next-generation sequencing delineates clonal origins and informs therapeutic strategies in acute lymphoblastic leukemia and histiocytic sarcoma. JCO Precision Oncology2019.
Gunz FW, Gunz JP, Veale AM, Chapman CJ, Houston IB. Familial leukaemia: a study of 909 families. Scand J Haematol. 1975;15(2):117–31.
Kraft IL, Godley LA. Identifying potential germline variants from sequencing hematopoietic malignancies. Blood. 2020;136(22):2498–506.
Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. 2015;373(24):2336–46.
Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27(5):658–70.
Sébert M, Passet M, Raimbault A, Rahmé R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. 2019;134(17):1441–4.
Bousquet M, Broccardo C, Quelen C, Meggetto F, Kuhlein E, Delsol G, et al. A novel PAX5-ELN fusion protein identified in B-cell acute lymphoblastic leukemia acts as a dominant negative on wild-type PAX5. Blood. 2007;109(8):3417–23.
Coyaud E, Struski S, Prade N, Familiades J, Eichner R, Quelen C, et al. Wide diversity of PAX5 alterations in B-ALL: a Groupe Francophone de Cytogenetique Hematologique study. Blood. 2010;115(15):3089–97.
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–31.
Duployez N, Jamrog LA, Fregona V, Hamelle C, Fenwarth L, Lejeune S, et al. Germline PAX5 mutation predisposes to familial B acute lymphoblastic leukemia. Blood. 2020.
Roloff GW, Godley LA, Drazer MW. Assessment of technical heterogeneity among diagnostic tests to detect germline risk variants for hematopoietic malignancies. Genet Med. 2020.
Li FP, Fraumeni JF. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–52.
Zebisch A, Lal R, Müller M, Lind K, Kashofer K, Girschikofsky M, et al. Acute myeloid leukemia with TP53 germ line mutations. Blood. 2016;128(18):2270–2.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52.
Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125(5):552–9.
Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122–37.
Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36(6):591–9.
Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer. 2016;122(2):304–11.
McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17(9):513–27.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Frisch A, Ofran Y. How I diagnose and manage Philadelphia chromosome-like acute lymphoblastic leukemia. Haematologica. 2019;104(11):2135–43.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15.
Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524–39.
Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12(6):344–57.
Churchman ML, Mullighan CG. Ikaros: Exploiting and targeting the hematopoietic stem cell niche in B-progenitor acute lymphoblastic leukemia. Exp Hematol. 2017;46:1–8.
Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood. 2019;134(16):1351–5.
Feurstein S, Godley LA. Germline ETV6 mutations and predisposition to hematological malignancies. Int J Hematol. 2017;102(6):189–195.
Gocho Y, Yang JJ. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood. 2019;134(10):793–797.
Stieglitz E, Loh ML. Genetic predispositions to childhood leukemia. Ther Adv Hematol. 2013;4(4):270–90.
Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112(8):1392–7.
de Miranda NF, Björkman A, Pan-Hammarström Q. DNA repair: The link between primary immunodeficiency and cancer. Ann N Y Acad Sci. 2011;1246:50–63.
Obrochta E, Godley LA. Identifying patients with genetic predisposition to acute myeloid leukemia. Best Pract Res Clin Haematol. 2018;31(4):373–378.
Author information
Authors and Affiliations
Contributions
G.W.R. and E.A.G conceived and designed the article. A.A., A.R.H, and G.W.R. performed the primary literature review. All authors contributed to the writing of the manuscript. All authors approve of the final manuscript and are accountable for the work.
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Griffiths has received honoraria/advisory board payments from Alexion Pharmaceuticals, Celgene/BMS, AbbVie, and Novartis. EAG has also received institutional research funding from Genentech Inc., Appelis pharmaceuticals, Celldex Therapeutics, and Celgene/BMS. The other authors declare no conflicts of interest/competing interests.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Acute Lymphocytic Leukemias
Rights and permissions
About this article

Cite this article
Aleem, A., Haque, A.R., Roloff, G.W. et al. Application of Next-Generation Sequencing-Based Mutational Profiling in Acute Lymphoblastic Leukemia. Curr Hematol Malig Rep 16, 394–404 (2021). https://doi.org/10.1007/s11899-021-00641-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-021-00641-5