
Abstract
Purpose of Review
Smoldering multiple myeloma (SMM) is defined as an asymptomatic clonal proliferation of pre-malignant plasma cells and an increased risk of progression to multiple myeloma (MM) relative to monoclonal gammopathy of undetermined significance. Whether patients with SMM should be treated prior to development of symptomatic disease is fiercely debated and is a highly active area of research.
Recent Findings
The ECOG E3A06 study demonstrated that early treatment with lenalidomide significantly reduced the risk of progression to MM compared to observation in patients with high risk SMM. The IMWG recently validated a risk stratification model to include cytogenetics and a personalized risk calculator for individual patients. Beyond this, molecular genomic aberrations and immunological phenomena that promote progression from asymptomatic disease to MM have been recently characterized and may help to more precisely identify patients who are most suitable for early intervention.
Summary
As highly effective and tolerable therapies for plasma cell disorders evolve, the field is approaching a paradigm shift that involves the adoption of intervention for patients with SMM who are at high risk for progression to symptomatic myeloma in order to prevent morbidity and mortality. This review highlights our current understanding of the biology of patients with SMM, clarifies the rationale for early intervention, and summarizes early results of various treatment strategies for patients with high-risk smoldering myeloma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy [1] and is characterized by the development of renal insufficiency, lytic bone disease, anemia, and hypercalcemia. Although therapeutic breakthroughs including immunomodulatory agents, proteasome inhibitors, and monoclonal antibodies have dramatically improved survival for patients with MM, early mortality rates remain unacceptably high [2] and many patients suffer permanent injuries including debilitating fractures and irreversible kidney disease that often require substantial supportive care [3, 4]. As such, identifying strategies that may prevent or reduce morbidity and mortality from MM is an important area of ongoing research. Elegant studies have demonstrated that nearly all cases of MM are preceded by monoclonal gammopathy of undetermined significance (MGUS) and/or smoldering multiple myeloma (SMM), precursor plasma cell neoplasms with a highly variable time course preceding the onset of symptoms related to MM [5, 6]. One strategy to potentially prevent complications and mortality from MM is to identify patients who may be appropriate for treatment before the development of any symptoms. A proper understanding of the biology of precursor plasma cell conditions and the risk posed by various clinical, genetic, and immunological factors for progression to myeloma is essential to identifying these patients.
Current Definitions and Risk of Progression
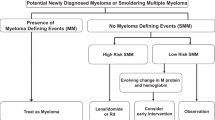
MGUS is characterized by a low quantity of clonal plasma cells within the bone marrow (< 10%) and low-to-moderate production of monoclonal protein (< 3 g/dL) in the absence of symptoms related to MM, lymphoma, or amyloidosis. SMM represents a more advanced precursor state with ≥ 10% plasma cells and/or ≥ 3 g/dL of monoclonal protein without any of the CRAB criteria (hypercalcemia, renal insufficiency, anemia, and lytic bone disease) or myeloma defining events as outlined in the most recent International Myeloma Working Group (IMWG) diagnostic criteria (serum free light chain (sFLC) ratio ≥ 100, ≥ 60% bone marrow plasma cells (BMPC), or > 1 focal lesion on MRI) [7]. Patients diagnosed with MGUS have a low risk of progression to MM of approximately 1% per year, with somewhat higher risk among those with monoclonal protein ≥ 1.5 g/dL and/or an abnormal free light chain ratio, with a risk of progression from MGUS to MM that remains fairly consistent over many years [8]. In contrast, patients with SMM have a much higher likelihood of progression, estimated at approximately 10% per year for the first 5 years after diagnosis [9]. Among patients with SMM, the risk of progression is highly variable, and thus, risk stratification models have been designed in order to estimate patients’ risk of progression to MM.
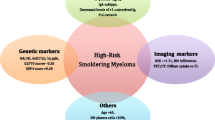
In current practice, the most commonly used risk stratification models are based upon clinical factors that estimate overall disease burden as a surrogate for malignant potential. These models have some overlap, but with some important differences, and they are summarized in Table 1. The original model proposed by the Mayo Clinic described three risk groups, based on the diagnostic criteria for SMM [9]. This was subsequently refined to include the serum-free light chain ratio as an important prognostic factor, and risk was determined according to whether patients met one, two, or three of the following criteria: monoclonal protein ≥ 3 g/dL, BMPC ≥ 10%, and sFLC ratio of ≥ 8 or ≤ 0.125 [10]. Ten years later, the same group proposed a new model with adjustment of the cutoff points to BMPC ≥ 20%, M-protein of 2 g/dL, and involved-to-uninvolved sFLC ratio of ≥ 20. This has become known as the “20/2/20” model and was shown to have a superior ability to predict progression to MM compared to the Mayo 2008 criteria [11]. Patients meeting all three of the original criteria, or just two of the “20/2/20” criteria, are considered to have high risk SMM, with approximately 50% risk of progression to MM within 2 years of diagnosis. The appeal of these models is that they are based upon tests that are routinely obtained in clinic, easily interpreted, widely available, reproducible, and serum biomarkers can be followed noninvasively over time to monitor for “evolving” changes in paraprotein production, which has been shown to be an ominous sign for progression to MM [12,13,14]. However, not all patients show this “evolving” pattern before progressing to symptomatic disease, and a recent study showed that patients whose plasma cells did not express CD56 were more likely to progress without a rise in monoclonal protein [15]. On the contrary, some patients with high-risk SMM never go on to develop MM, but the ability to predict which of these high-risk SMM patients will remain stable over time remains elusive.
A striking pattern seen among the clinical risk models is that the highest risk of progression to MM is observed within the first 2 years of diagnosis regardless of risk category. As such, there are likely at least two biological entities that exist among patients with SMM—“high-volume MGUS” that follows a relatively indolent course and “pre-symptomatic myeloma” that is a true malignancy with impending development of symptomatic disease. Indeed, a study of patients with low-risk MGUS by conventional criteria found that 45% of patients who otherwise would not have required a bone marrow biopsy based on monoclonal protein quantity actually had ≥ 10% BMPCs and met the diagnostic criteria for SMM, but still had a risk of progression of ~ 2% per year, far lower than the average risk for SMM [16]. This highlights that while clinical models do a good job of estimating risk for the majority of patients, basing decisions on these models alone will certainly lead to under treatment of some SMM patients with “low-risk” disease who actually have “pre-symptomatic myeloma,” and over treatment of some SMM patients with “high-volume MGUS” who appear to be high risk. Avoiding the use of unnecessary and potentially toxic therapies that may harm quality of life and actually shorten survival of such SMM patients is imperative. However, with proper diagnosis and risk categorization, early treatment of patients with “pre-symptomatic myeloma” does have the potential to benefit patients, not only by reducing the likelihood of symptom development but also to potentially prolong survival. As such, implementing additional genomic and immunological information along with these clinical models will likely help to improve outcomes for all patients with SMM.
Genomics
MM is characterized by recurrent structural genetic changes and cytogenetic abnormalities (CA) that are acquired early in development of the plasma cell neoplasm and serve as important prognostic and predictive biomarkers [17, 18]. Most of the CA found among patients with MM have also been identified in patients with MGUS and SMM. Several of these, including t(4;14), del(17p), + 1q, and hyperdiploidy have been associated with an increased risk of progression from SMM to MM [19, 20]. In 2020, the IMWG introduced a SMM new risk model incorporating cytogenetic abnormalities and explored the relative risk of each factor on a continuous spectrum [21••]. In this model, patients meeting 3 of 4 high-risk features, including those defined by the “20/2/20” model along with abnormal cytogenetics [t(4;14), t(14;16), + 1q, and/or del(13q/− 13}, as assessed by FISH, were noted to have a 63.1% risk of progression to MM at 2 years, versus 45.5% for those with 2 criteria, 22.8% with 1 criterion, and only 6% with none of the above. Additionally, a risk calculator was derived that allowed for determination of risk based on the degree of abnormality among each of the criteria, thus providing a more precise means to estimate risk for individual patients. This calculator is summarized in Table 1, with high-risk patients having a 2-year risk of progression of 72.5%.
Beyond the recurrent CAs that are routinely evaluated among patients with plasma cell disorders, complex structural changes are becoming increasingly recognized as an important factor in determining outcomes among patients with MM, and now have shown promise in predicting progression to myeloma. A recent study using whole genome sequencing (WGS) identified three classes of complex structural variants among patients with MM—chromothripsis, templated insertions, and chromoplexy. In particular, chromothripsis—which is marked by multifocal chromosomal shattering and random rejoining of pieces—results in hundreds of new chromosome recombinations and copy number variation, and is associated with inferior survival in MM [22]. The same group recently performed a similar analysis among patients with SMM and found that while chromothripsis was uncommon in SMM, it was never seen among patients with stable disease [23]. Another study using high-throughput genomic analysis found that aneuploidies, especially deletions, as well as a higher “genomic scar score” were more common in patients progressing from SMM to MM compared to nonprogressors [24].
In addition to large structural changes, mutations at the genetic level are also found at high frequency among patients with plasma cell disorders. Several groups have demonstrated that the mutational landscape of patients with SMM is very similar to that of newly diagnosed MM [23, 25, 26•]. Among patients with SMM, several specific mutational patterns have emerged as highly predictive of progression to MM. Bustoros et al. determined that mutations within the mitogen-activated protein kinase (MAPK) pathway (KRAS, NRAS), DNA damage repair pathway (TP53, ATM), and MYC alterations were associated with high rates of progression to MM [26•]. Mutations within the MAPK pathway are among the most common in MM [27], and TP53 mutations are known to be one of the most important prognostic factors in MM, particularly when combined with del(17p) [28]. Mounting evidence suggests that MYC alterations almost universally signal high-risk disease among plasma cell disorders. The acquisition of MYC alterations triggers progression of MGUS to MM in mouse models [29, 30]. Multiple studies suggest that MYC alterations are inconsistent with stable precursor diseases [24, 31, 32], and MYC translocations have been associated with poor prognosis when identified among patients with MM [31, 33]. When evaluated in conjunction with clinical criteria, among patients within each of the “20/2/20” risk categories, patients with MAPK, DNA damage repair, or MYC alterations had higher risk of progression compared to those without these mutations [26•]. The APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) mutational signature has also been associated with inferior outcomes in MM and was recently identified as an important predictor for progression from SMM to MM as well [24].
While these data suggest that the acquisition of complex structural variation and aggressive mutational signatures may indicate that an asymptomatic plasma cell clone is likely to progress to symptomatic MM, this remains an incomplete characterization of all patients. Multiple studies have demonstrated that, in addition to dominant chromosomal and mutational changes, the subclonal genomic landscape frequently changes as disease progresses from SMM to MM [25, 34, 35]. Furthermore, patients who progressed without subclonal evolution often had high tumor burden at diagnosis or short time to progression, indicating that these changes may have already occurred in some patients with high-risk SMM at the time of diagnosis. These studies provide additional genomic evidence to support the theory that two distinct disease biologies within SMM exist, as mentioned previously. While some genomic abnormalities, such as MYC alterations, may be sufficient to drive progression to symptomatic MM, the complex interplay between the immune microenvironment and tumor cells may also be a key factor in determining the rate of progression and/or whether additional mutational burden is required.
Immunology
Clearly, incorporation of genomic information can help to refine risk stratification in SMM, but the complex interactions between the various immune subsets within the bone marrow microenvironment remain an important prognostic factors in addition to potentially inducing control of the pre-malignant clone. In fact, prior to the development of the original Mayo Clinic risk stratification scheme, the Grupo Espanol de Mieloma/Programa para el Tratamiento de Hemopatias Malignas (PETHEMA/GEM) developed the first published risk stratification model for SMM based on immunological parameters. In this model, high-risk criteria were defined as the presence of immunoparesis, defined as a reduction in at least one immunoglobulin class, and if ≥ 95% of the plasma cells in the bone marrow aspirate had an aberrant phenotype by flow cytometry [36]. Although this model was not widely incorporated after its publication, it has been validated and underscores the importance of the overall immunophenotype and interplay between the plasma cell neoplasm and immune microenvironment.
T cells are well described to be able to induce anti-myeloma responses among patients with MM [37]. Early immunological profiling determined that effector T cells mount a vigorous anti-myeloma effect to control pre-malignant plasma cell clones [38], and that progression of MM is characterized by deficiencies in adaptive immunity [39]. In the peripheral blood, among patients progressing to MM, there is a notable decrease in effector memory and cytotoxic T cells and increased Tγδ cells and adaptive NK cells [40].
Beyond T cells, a more extensive characterization of the immune landscape has been described in recent years. Compared to healthy controls, patients with plasma cell neoplasms have increased terminal effector T cell differentiation, but stem-like T cells that are identified at the MGUS stage are lost upon progression to MM [41]. Single-cell RNA sequencing of patients with MGUS, SMM, and MM has identified that patients who have advanced to SMM or MM have loss of memory cytotoxic T cells and MHC Class II dysregulation among monocytes in the bone marrow microenvironment, leading to T cell suppression and loss of control of the plasma cell neoplasm [42•]. Immune checkpoints also appear to be dysregulated among patients progressing from MGUS to MM [43]. Although one study demonstrated that the PD-1/PD-L1 phenotype is no different among patients with SMM or newly diagnosed MM, this becomes skewed in relapsed/refractor disease, suggesting that the T cell repertoire of patients with SMM may be more capable of recovering immunological control compared to the relapsed stage [44]. Several longitudinal studies are ongoing to characterize the feasibility of immune cell profiling to not only identify patients at high risk of progression but also potentially highlight opportunities for treatment to regain immunological control of the premalignant clone. Supporting this approach are data from a randomized study, in which patients with high-risk SMM were noted to have decreased expression of activated TH1 cells at baseline, but treatment with lenalidomide and dexamethasone treatment was able to restore T cell and NK cell phenotypes to those resembling healthy controls [45]. Clearly, immunological processes are critical to the overall control of SMM in the premalignant state, and strategies that optimize immune control are attractive, particularly for those who may be at high risk for early progression.
Treatment for Smoldering Myeloma
By definition, SMM is asymptomatic, and any discussion of treatment for an asymptomatic condition requires at least a basic understanding of the nuance regarding the concepts of treating pre-malignant conditions and asymptomatic malignancies. Early intervention for pre-malignant conditions introduces lead time bias that can artificially inflate the apparent benefit of early intervention, so results need to be interpreted with caution. Additionally, the risk:benefit ratio of early intervention to reduce cancer-related mortality coupled with the possibility of over diagnosis and potential harms of treatment need to be carefully examined. On one end of the spectrum, polypectomy during colonoscopy is universally recommended because of the known risk of progression of pre-malignant polyps to colon cancer, high cure rate with polypectomy, and very low risk of the endoscopic procedure. On the contrary, despite being a true malignancy, expert consensus is to avoid treating patients with asymptomatic indolent lymphoma not meeting Groupe d’Etude des Lymphomes Folliculaires (GELF) criteria, based on the lack of a survival benefit to early intervention [46], low risk of complications with noninvasive surveillance, and substantial toxicity of treatment.
For decades, since its initial description by Dr. Robert Kyle in 1980 [47], “observation only” was the sine qua non for smoldering myeloma, a recommendation that undoubtedly reflected the original understanding of SMM as an intermediate stage of progression from MGUS to MM rather than a basket term including some patients with “high-volume MGUS” and others with “pre-symptomatic myeloma.” Reinforcing the assumption, a number of trials were completed in the 1990s with available agents that failed to slow progression to symptomatic myeloma. One of the largest trials randomized 145 patients with asymptomatic myeloma to either 6 cycles of melphalan and prednisone or treatment at time of development of symptoms, and found no difference in overall survival, with the best survival among the 32 patients who never received treatment [48]. Agents such as bisphosphonates, either singly or in combination with thalidomide [49,50,51], also failed to change overall survival, although the addition of thalidomide did slow progression to MM.
The arrival of novel agents rekindled the interest in treating SMM, due to their greater efficacy and manageable side effects even on a longer-term basis. Clinical trials examining the utility of bortezomib in SMM produced inconclusive results and were abandoned [52]. More recently, lenalidomide became the obvious candidate to consider for SMM patients due to experience gained with this drug in long-term MM maintenance and treatment trials [53, 54]. In 2007, the PETHEMA group initiated the QUIREDEX study, a randomized, open-label trial of lenalidomide plus dexamethasone (Rd) versus observation in high-risk SMM, defined as either > 10% PCs and > 3g/dL, or if only one criterion was present, high risk by virtue of the PETHEMA risk classification, i.e., aberrant flow and/or immunoparesis. Patients in the Rd group were administered nine 4-week induction cycles (lenalidomide 25 mg/day on days 1–21, plus dexamethasone 20 mg per day on days–1–4 and days 12–15), followed by maintenance with lenalidomide at 10 mg/day on days 1–21 of each 28-day cycle for up to 2 years. The protocol permitted the reintroduction of low-dose dexamethasone (20 mg on days 1–4 of each cycle) in subjects who developed asymptomatic biological progression during the maintenance phase. By contrast, patients in the control (observation) group were not permitted to receive treatment until progression to symptomatic disease. Ultimately, 119 patients were enrolled, with those patients receiving Rd showing a progression free survival (PFS) advantage (not reached vs. 23 months; HR 0.18) as well as a 3-year overall survival (OS) advantage of 94% vs. 80 (HR 0.31) [55, 56]. However, this approach was not immediately embraced for several key reasons. First, advanced imaging (MRI, PET-CT, whole-body low-dose CT) to detect skeletal lesions to exclude active MM cases was not required, and the rapid progression to MM seen early on in the observation arm implies that some patients with active MM were inadvertently included. In addition, patients on the R maintenance who began to show signs of biochemical progression had dexamethasone reintroduced, altering the study design from true limited intervention vs. observation. Moreover, because a combination regimen was used, the incremental value of adding lenalidomide could not be clearly delineated. Nonetheless, the study achieved an important milestone and clearly spurred the development of additional trials for SMM.
To date, the largest randomized SMM trial is ECOG-E3A06, which enrolled SMM patients to either indefinite lenalidomide (R) monotherapy or observation. This trial did mandate advanced imaging to rule out lytic lesions but was amended to include low-risk SMM and diagnosis within 5 years to enhance accrual, and included patients with intermediate- or high-risk SMM, with PFS (defined as the time since randomization to the development of symptomatic end-organ damage—osteolytic bone lesions, acute renal failure, etc.) as the primary endpoint. A total of 224 patients were enrolled, with 182 randomized, after an initial lead-in phase to establish safety. Subjects receiving R experienced a median 3-year PFS of 91% vs. 66% for the observation arm (HR 0.28, P = .002), despite a relatively low overall response rate (ORR) of 50% [57••]. The PFS advantage to early intervention with R monotherapy was most notable for patients who were categorized as high risk by the Mayo 20-2-20 criteria. The treatment was not associated with a measurable decline in quality-of-life scores, although 40% of patients on the R arm came off treatment due to adverse events, and the median time on R was approximately 2 years. Unlike the QUIREDEX study, so far, no survival benefit has been observed with continuous R therapy, with only six 6 deaths reported (2 in R arm vs. 4 with observation; HR 0.46 (95% CI 0.08–2.53).
The results of E3A06 and QUIREDEX trials, as well as the availability of newer additional agents, have contributed to the burgeoning number of SMM trials currently underway. The treatment paradigm for these trials can be roughly divided into two groups: limited therapy designed to halt progression to MM and perhaps return patients to an “MGUS-like state,” versus those using aggressive MM-style therapy to induce deep remissions and high rates of MRD negativity, with a goal of eradicating the premalignant clone(s) and/or resulting in a functional cure for many patients.
Examples of the aggressive approach are growing. Investigators from the National Cancer Institute recently updated and extended results from their trial of carfilzomib, lenalidomide, and dexamethasone (KRd) for 8 cycles, followed by 2years of lenalidomide maintenance [58, 59]. Their most recent publication includes 54 patients with high-risk SMM, defined in a number of different ways, treated with this regimen. The overall response rate was 100%, with a CR rate of 72.7%. However, 50% of patients experienced at least one grade 3 adverse event. At a 5-year landmark, the PFS was 90%, and there were no treatment related deaths on this study. The phase II GEM-CESAR trial represents the most aggressive approach to date for high-risk SMM, defined by the PETHEMA definition, with the use of advanced imaging to rule out lytic lesions in this particular trial. A total of 90 patients were enrolled and treated with 6 cycles of KRd followed by autologous transplant, 2 more cycles of KRd, and up to 2 years of lenalidomide maintenance. Preliminary results from this trial [60, 61] show a CR rate of 70%, with an MRD negative rate of 60%. Three out of 90 patients have died, with 1 death linked to treatment related toxicity. It is also important to note that, as in the previous Spanish trial, approximately 30% of patients in the GEM-CESAR study would meet the current IMWG definition of active MM. In the United States, the ASCENT trial (Aggressive Smoldering Curative Approach Evaluating Novel Therapies) is a phase II, single-arm study that is enrolling high-risk SMM patients and treating them with a four-drug regimen (carfilzomib, daratumumab, dexamethasone, and lenalidomide) for a total of 24 monthly cycles, again with the aim of producing deep remissions and arresting progression to active MM (NCT03289299).
The other approach to SMM treatment focuses more on limited, less intensive therapy, with the goal of slowing or halting progression to active MM, and limiting toxicity in a group of subjects that are by definition asymptomatic. Both the QUIREDEX and E3A06 studies typify this approach. The CENTAURUS study evaluated single-agent daratumumab (Dara) in several dosing strategies for treatment of SMM. Dara resulted in an ORR of 37.5–56.1%, CR rates of 0–9.8%, and a 24-month PFS of 75.6–87.8% [62]. The co-primary endpoints of CR > 15% was not met. Two ongoing studies—the AQUILA trial (NCT03301220), which enrolled 390 patients and randomized patients to receive subcutaneous Dara vs. observation, and the Phase III DETER-SMM trial (EAA173, NCT03937635), which will randomize 288 patients with high-risk SMM as defined by the 20/2/20 criteria to Dara-Rd versus Rd—will help to further explore the optimal preventive approach to high-risk SMM. It is noteworthy that the DETER trial offers treatment in both study arms, an acknowledgment that high-risk SMM patients should no longer be randomized to placebo or observation. The AQUILA trial eligibility enrolled patients defined as high risk with less stringent criteria than DETER, specifying that subjects SMM within 5 years of diagnosis, 10% PC, and only one of the following criteria: M spike > 3 g/dL, IgA SMM, immunoparesis, or FLC ratio > 8 but < 100. Results are expected in the fourth quarter of 2021.patients. A large multinational trial will evaluate the effectiveness of another anti-CD 38 antibody, isatuximab, in combination with Rd versus Rd alone (NCT02916771).
A growing area of interest in the field of SMM treatment involves immunotherapeutic approaches. Although studies using checkpoint inhibitors in MM have been disappointing [63, 64], the T cell repertoire of patients with precursor plasma cell neoplasms may be better suited for targeting by these approaches, as mentioned above. A stringent CR was seen in a patient with high-risk genetics in an early report evaluating pembrolizumab in SMM [65], and a pilot study of the 4-peptide directed PVX-410 vaccine, in combination with lenalidomide, produced sustained increases in CD8+ memory T cells, and was well tolerated [66]. Other “personalized” vaccine trials are underway, and may reveal the importance of this approach in SMM.
All of this activity begs the question of whether SMM patients should be treated routinely outside of a clinical trial. One caveat regarding the completed trials is the lack of uniform entrance criteria, which hampers the interpretation and applicability of any one trial’s results to the larger SMM patient population. The recently published IMWG SMM risk scoring system [21••] will likely set the new eligibility standard for future trials, particularly given how well low-risk SMM patients fare without intervention. Some investigators have come out in favor of treatment off-trial for SMM [67, 68], although others have promoted restraint and asked for a more dynamic assessment of SMM risk as it evolves over time, before committing to treatment [69]. Furthermore, some have proposed that early intervention should be avoided until a clear benefit in overall survival or quality of life is demonstrated in large, randomized controlled trials [70]. All experts agree that any patient diagnosed with SMM needs thorough evaluation to assure that the individual does not meet the current definition of MM and require immediate treatment. Patients with low-risk SMM should be followed expectantly. It is prudent to follow newly diagnosed SMM patients closely to make sure that they do not have rapidly increasing monoclonal protein levels and/or FLC ratios that would change risk assessment. In large part due to the rapidly changing and highly controversial landscape of SMM management, we strongly recommend referring any SMM patient for one of the numerous clinical trials underway as outlined in Table 2. Undoubtedly, the development of newer diagnostic platforms will allow for even better discrimination of risk of SMM progression and recommendations for intervention will continue to evolve. In conjunction with this, as the effect of interventions and patient outcomes are further defined by ongoing clinical trials, a much clearer understanding of how, when, and which patients with SMM should be treated will likely become apparent in the near future.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
American Cancer Society. Cancer facts & figures 2019. 2019.
Costa LJ, Bal S, Chhabra S. Population-level trends in early mortality and overall survival of patients with multiple myeloma. Are we facing stagnation? Blood. 2019;134(Supplement_1):4760. https://doi.org/10.1182/blood-2019-121786.
Terpos E, Morgan G, Dimopoulos MA, Drake MT, Lentzsch S, Raje N, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol. 2013;31(18):2347–57. https://doi.org/10.1200/JCO.2012.47.7901.
Dimopoulos MA, Sonneveld P, Leung N, Merlini G, Ludwig H, Kastritis E, et al. International Myeloma Working Group recommendations for the diagnosis and management of myeloma-related renal impairment. J Clin Oncol. 2016;34(13):1544–57. https://doi.org/10.1200/JCO.2015.65.0044.
Landgren O, Kyle R, Pfeiffer R. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113:5412–7. https://doi.org/10.1182/blood-2008-12-194241.
Weiss B, Abadie J, Verma P. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–22. https://doi.org/10.1182/blood-2008-12-195008.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–48. https://doi.org/10.1016/S1470-2045(14)70442-5.
Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–9. https://doi.org/10.1056/NEJMoa1709974.
Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–90. https://doi.org/10.1056/NEJMoa070389.
Dispenzieri A, Kyle RA, Katzmann JA, Therneau TM, Larson D, Benson J, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111(2):785–9. https://doi.org/10.1182/blood-2007-08-108357.
Lakshman A, Rajkumar SV, Buadi FK, Binder M, Gertz MA, Lacy MQ, et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018;8(6):59. https://doi.org/10.1038/s41408-018-0077-4.
Rosinol L, Blade J, Esteve J, Aymerich M, Rozman M, Montoto S, et al. Smoldering multiple myeloma: natural history and recognition of an evolving type. Br J Haematol. 2003;123(4):631–6. https://doi.org/10.1046/j.1365-2141.2003.04654.x.
Ravi P, Kumar S, Larsen JT, Gonsalves W, Buadi F, Lacy MQ, et al. Evolving changes in disease biomarkers and risk of early progression in smoldering multiple myeloma. Blood Cancer J. 2016;6(7):e454. https://doi.org/10.1038/bcj.2016.65.
Fernandez de Larrea C, Isola I, Pereira A, Cibeira MT, Magnano L, Tovar N, et al. Evolving M-protein pattern in patients with smoldering multiple myeloma: impact on early progression. Leukemia. 2018;32(6):1427–34. https://doi.org/10.1038/s41375-018-0013-4.
Notarfranchi L, Vescovini R, Segreto R, Bonomini S, Storti P, Marchica V, et al. Short-term risk for progression in patients with smoldering multiple myeloma: the impact of CD56 expression. Blood. 2020;136(Supplement 1):11. https://doi.org/10.1182/blood-2020-139214.
Bustoros M, Kastritis E, Sklavenitis-Pistofidis R. Bone marrow biopsy in low-risk monoclonal gammopathy of undetermined significance reveals a novel smoldering multiple myeloma risk group. Am J Hematol. 2019;94:E146–9. https://doi.org/10.1002/ajh.25441.
Kumar SK, Rajkumar SV. The multiple myelomas—current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol. 2018;15(7):409–21. https://doi.org/10.1038/s41571-018-0018-y.
Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front Immunol. 2019;10:1121. https://doi.org/10.3389/fimmu.2019.01121.
Neben K, Jauch A, Hielscher T, Hillengass J, Lehners N, Seckinger A, et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 2013;31(34):4325–32. https://doi.org/10.1200/JCO.2012.48.4923.
Rajkumar SV, Gupta V, Fonseca R, Dispenzieri A, Gonsalves WI, Larson D, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27(8):1738–44. https://doi.org/10.1038/leu.2013.86.
•• Mateos M-V, Kumar S, Dimopoulos MA, González-Calle V, Kastritis E, Hajek R, et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020;10(10):102. https://doi.org/10.1038/s41408-020-00366-3This is the most recent and validated risk stratification model for SMM and has also incorporated cytogenetics into the model, likely indicating the beginning of a trend to incorporate genomic data into SMM risk stratification.
Rustad EH, Yellapantula VD, Glodzik D, Maclachlan KH, Diamond B, Boyle EM, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov. 2020;1:258–73. https://doi.org/10.1158/2643-3230.Bcd-20-0132.
Oben B, Froyen G, Maclachlan KH, Zheng-Lin B, Yellapantula V, Abascal F, et al. Whole-genome sequencing reveals evidence of two biologically and clinically distinct entities: progressive versus stable myeloma precursor disease. Blood. 2020;136(Supplement 1):47–8. https://doi.org/10.1182/blood-2020-136403.
Aktas-Samur A, Fulciniti M, Derebail S, Szalat R, Parmigiani G, Corre J, et al. High throughput genomic analysis identifies low-risk smoldering multiple myeloma. Blood. 2020;136(Supplement 1):2. https://doi.org/10.1182/blood-2020-139066.
Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. 2018;9(1):3363. https://doi.org/10.1038/s41467-018-05058-y.
• Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford AJ, et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J Clin Oncol. 2020. https://doi.org/10.1200/JCO.20.00437 JCO2000437. This paper detailed the most important mutations that predict progression of SMM to MM.
Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–16. https://doi.org/10.1038/s41375-018-0037-9.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2018;33:159–70. https://doi.org/10.1038/s41375-018-0196-8.
Affer M, Chesi M, Chen WG, Keats JJ, Demchenko YN, Roschke AV, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28(8):1725–35. https://doi.org/10.1038/leu.2014.70.
Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13(2):167–80. https://doi.org/10.1016/j.ccr.2008.01.007.
Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000;97(1):228–33. https://doi.org/10.1073/pnas.97.1.228.
Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia. 2020;34(1):322–6. https://doi.org/10.1038/s41375-019-0543-4.
Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4(3):e191. https://doi.org/10.1038/bcj.2014.13.
Boyle EM, Davies FE, Deshpande S, Tytarenko RG, Ashby C, Wang Y, et al. Analysis of the sub-clonal structure of smoldering myeloma over time provides a new means of disease monitoring and highlights evolutionary trajectories leading to myeloma. Blood. 2019;134(Supplement_1):4333. https://doi.org/10.1182/blood-2019-126679.
Merz M, Hielscher T, Schult D, Mai EK, Raab MS, Hillengass J, et al. Cytogenetic subclone formation and evolution in progressive smoldering multiple myeloma. Leukemia. 2020;34(4):1192–6. https://doi.org/10.1038/s41375-019-0634-2.
Perez-Persona E, Vidriales MB, Mateo G, Garcia-Sanz R, Mateos MV, de Coca AG, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–92. https://doi.org/10.1182/blood-2007-05-088443.
Dhodapkar MV, Krasovsky J, Olson K. T cells from the tumor microenvironment of patients with progressive myeloma can generate strong, tumor-specific cytolytic responses to autologous, tumor-loaded dendritic cells. Proc Natl Acad Sci U S A. 2002;99(20):13009–13. https://doi.org/10.1073/pnas.202491499.
Dhodapkar MV, Krasovsky J, Osman K, Geller MD. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J Exp Med. 2003;198(11):1753–7. https://doi.org/10.1084/jem.20031030.
Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197(12):1667–76. https://doi.org/10.1084/jem.20021650.
Termini R, Terpos E, Pérez A, Jelinek T, Kokkali N-A, Bargay J, et al. Longitudinal immunogenomic profiling of tumor and immune cells for minimally-invasive monitoring of smoldering multiple myeloma (SMM): the immunocell study. Blood. 2020;136(Supplement 1):1–2. https://doi.org/10.1182/blood-2020-136251.
Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. 2019;5. https://doi.org/10.1172/jci.insight.127807.
• Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Can. 2020. https://doi.org/10.1038/s43018-020-0053-3This paper highlights key aspects of the immunological milieu of myeloma precursor states.
Dhodapkar MV, Sexton R, Das R, Dhodapkar KM, Zhang L, Sundaram R, et al. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood. 2015;126(22):2475–8. https://doi.org/10.1182/blood-2015-03-632919.
Costa F, Vescovini R, Notarfranchi L, Storti P, Marchica V, Dalla Palma AB, et al. PD-L1/PD-1 pattern of distribution within bone marrow microenvironment cells in patients with smoldering myeloma and active multiple myeloma. Blood. 2020;136(Supplement 1):49–50. https://doi.org/10.1182/blood-2020-139275.
Paiva B, Mateos MV, Sanchez-Abarca LI, Puig N, Vidriales MB, Lopez-Corral L, et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: a longitudinal analysis. Blood. 2016;127(9):1151–62. https://doi.org/10.1182/blood-2015-10-662320.
Brice P, Bastion Y, Lepage E, Brousse N, Haïoun C, Moreau P, et al. Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d’Etude des Lymphomes Folliculaires. Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 1997;15(3):1110–7. https://doi.org/10.1200/jco.1997.15.3.1110.
Kyle RA, Greipp PR. Smoldering multiple myeloma. N Engl J Med. 1980;302(24):1347–9. https://doi.org/10.1056/NEJM198006123022405.
Riccardi A, Mora O, Tinelli C, Valentini D, Brugnatelli S, Spanedda R, et al. Long-term survival of stage I multiple myeloma given chemotherapy just after diagnosis or at progression of the disease: a multicentre randomized study. Br J Cancer. 2000;82(7):1254–60. https://doi.org/10.1054/bjoc.1999.1087.
Witzig TE, Laumann KM, Lacy MQ, Hayman SR, Dispenzieri A, Kumar S, et al. A phase III randomized trial of thalidomide plus zoledronic acid versus zoledronic acid alone in patients with asymptomatic multiple myeloma. Leukemia. 2013;27(1):220–5. https://doi.org/10.1038/leu.2012.236.
D'Arena G, Gobbi PG, Broglia C, Sacchi S, Quarta G, Baldini L, et al. Pamidronate versus observation in asymptomatic myeloma: final results with long-term follow-up of a randomized study. Leuk Lymphoma. 2011;52(5):771–5. https://doi.org/10.3109/10428194.2011.553000.
Musto P, Petrucci MT, Bringhen S, Guglielmelli T, Caravita T, Bongarzoni V, et al. A multicenter, randomized clinical trial comparing zoledronic acid versus observation in patients with asymptomatic myeloma. Cancer. 2008;113(7):1588–95. https://doi.org/10.1002/cncr.23783.
Zangari M, Berno T, Salama ME, Sana S, Talamo G, Pena K, et al. Effect of low dose bortezomib on bone formation in smoldering myeloma patients. Blood. 2013;122(21):3204. https://doi.org/10.1182/blood.V122.21.3204.3204.
McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–81. https://doi.org/10.1056/NEJMoa1114083.
McCarthy PL, Holstein SA, Petrucci MT, Richardson PG, Hulin C, Tosi P, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35(29):3279–89. https://doi.org/10.1200/jco.2017.72.6679.
• Mateos MV, Hernandez MT, Giraldo P, de la Rubia J, de Arriba F, Lopez Corral L, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438–47. https://doi.org/10.1056/NEJMoa1300439This is the first randomized phase III trial to show a PFS and OS benefit in treating SMM with lenalidomide and steroids.
Mateos MV, Hernandez MT, Giraldo P, de la Rubia J, de Arriba F, Corral LL, et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17(8):1127–36. https://doi.org/10.1016/S1470-2045(16)30124-3.
•• Lonial S, Jacobus S, Fonseca R, Weiss M, Kumar S, Orlowski RZ, et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. J Clin Oncol. 2020;38(11):1126–37. https://doi.org/10.1200/jco.19.01740This study confirmed the PFS benefit of lenalidomide in the treatment of SMM and serves as a benchmark for future studies.
Mailankody S, Kazandjian D, Korde N, Roschewski M, Manasanch E, Bhutani M, et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv. 2017;1(22):1911–8. https://doi.org/10.1182/bloodadvances.2017005934.
Kazandjian D, Hill E, Morrison C, Dew A, Korde N, Mailankody S, et al. Treatment of high risk (HR) smoldering multiple myeloma (SMM) with carfilzomib, lenalidomide, and dexamethasone (KRd) followed by lenalidomide maintenance (-R): a phase 2 clinical and correlative study. Blood. 2020;136(Supplement 1):43–5. https://doi.org/10.1182/blood-2020-136148.
Puíg N, Contreras T. Analysis of treatment efficacy in the GEM-CESAR trial for high-risk smoldering multiple myeloma patients: comparison between the standard and IMWG MRD criteria and QIP-MS including FLC (QIP-FLC-MS). ASCO Virtual Sci Program: American Society of Clinical Oncology. 2020;38(15_suppl):8512.
Mateos M-V, Martinez-Lopez J, Rodriguez Otero P, Gonzalez-Calle V, Gonzalez MS, Oriol A, et al. Curative strategy (GEM-CESAR) for high-risk smoldering myeloma (SMM): carfilzomib, lenalidomide and dexamethasone (KRd) as induction followed by HDT-ASCT, consolidation with Krd and maintenance with Rd. Blood. 2019;134(Supplement_1):781. https://doi.org/10.1182/blood-2019-125204.
Landgren CO, Chari A, Cohen YC, Spencer A, Voorhees P, Estell JA, et al. Daratumumab monotherapy for patients with intermediate-risk or high-risk smoldering multiple myeloma: a randomized, open-label, multicenter, phase 2 study (CENTAURUS). Leukemia. 2020;34(7):1840–52. https://doi.org/10.1038/s41375-020-0718-z.
Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6(9):e448–e58. https://doi.org/10.1016/S2352-3026(19)30109-7.
Mateos M-V, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6(9):e459–e69. https://doi.org/10.1016/S2352-3026(19)30110-3.
Manasanch EE, Han G, Mathur R, Qing Y, Zhang Z, Lee H, et al. A pilot study of pembrolizumab in smoldering myeloma: report of the clinical, immune, and genomic analysis. Blood Adv. 2019;3(15):2400–8. https://doi.org/10.1182/bloodadvances.2019000300.
Nooka AK, Wang ML, Yee AJ, Kaufman JL, Bae J, Peterkin D, et al. Assessment of safety and immunogenicity of PVX-410 vaccine with or without lenalidomide in patients with smoldering multiple myeloma: a nonrandomized clinical trial. JAMA Oncol. 2018;4(12):e183267. https://doi.org/10.1001/jamaoncol.2018.3267.
Mateos M-V, González-Calle V. Timing of treatment of smoldering myeloma: early treatment. Blood Adv. 2018;2(21):3045–9. https://doi.org/10.1182/bloodadvances.2018021220.
Kapoor P, Rajkumar SV. Smoldering multiple myeloma: to treat or not to treat. Cancer J. 2019;25(1):65–71. https://doi.org/10.1097/ppo.0000000000000350.
Fonseca R, Gonzalez-Velez M. Treatment of smoldering multiple myeloma: expectant observation should still be the standard. Am Soc Clin Oncol Educ Book. 2020;40:364–70. https://doi.org/10.1200/EDBK_280179.
Goodman AM, Kim MS, Prasad V. Persistent challenges with treating multiple myeloma early. Blood. 2020;137:456–8. https://doi.org/10.1182/blood.2020009752.
Author information
Authors and Affiliations
Contributions
T.M.S. and N.S.C. contributed equally to the literature review and writing of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
T.M.S. has no conflicts of interest or other disclosures. N.S.C. has received research funding from Cellectar.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Multiple Myeloma
Rights and permissions
About this article

Cite this article
Schmidt, T.M., Callander, N.S. Progress in the Management of Smoldering Multiple Myeloma. Curr Hematol Malig Rep 16, 172–182 (2021). https://doi.org/10.1007/s11899-021-00623-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-021-00623-7