
Abstract
Purpose of Review
Peripheral T cell lymphoma is a rare heterogeneous group of diseases which are characterized by poor outcomes to treatment and short overall survival. In the past decade, several new therapies targeting T cell biology have been approved in the relapsed setting. These new therapies, such as pralatrexate, romidepsin, belinostat, and brentuximab vedotin, have begun to make their way into practice. Despite these advances, outcomes have not changed dramatically. In recent years, efforts have been made to incorporate these new therapies into combination strategies to treat this challenging disease entity. Herein we will review some of the latest developments.
Recent Findings
With the new WHO classification, discrete entities of PTCL are now being identified by molecular and phenotypic markers. This new classification is critical to our ability to define disease entities which may respond to certain classes of targeted therapy. Some such mutations include genes controlling epigenetics (TET2, IDH2, DNMT3A, RHOA, CD28). As such, epigenetic therapies such as histone deacetylase (HDAC) inhibitors have become the platform to which other novel therapies or chemotherapy has been added. Early phase clinical studies have demonstrated that combination therapy with romidepsin plus other agents known to have activity in T cell lymphoma have enhanced clinical benefit for this group of diseases. In addition, the antibody drug conjugate, brentuximab vedotin has been shown to have potent activity in T cell lymphomas expressing CD30. This drug is being studied as well with other targeted therapies and chemotherapy in an effort to improve response rates and progression-free survival.
Summary
Although T cell lymphomas remain a highly challenging group of diseases to treat, new efforts to leverage drugs that discretely target the biology that drives T cell lymphomagenesis in combination provide hope that improved outcomes may be realized in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2016, the World Health Organization (WHO) classified 28 subtypes of peripheral T cell lymphomas (PTCL), highlighting the extreme cytological and phenotypic heterogeneity of this rare group of diseases affecting post-thymic T cells [1••]. PTCL make up only 5–10% of all lymphoid neoplasms, making them a challenging group of diseases to study [2, 3].
Although the biology of PTCL is varied, there have been advancements in revealing the mechanisms of their malignant transformation. Anaplastic large cell lymphoma (ALCL) typically expresses CD30, a transmembrane glycoprotein member of the tumor necrosis factor receptor family [4]. There are three distinct variants of ALCL: systemic, cutaneous and breast implant associated subtypes. Systemic ALCL is further distinguished by ALK rearrangements which occur following the translocation of chromosomes 2 and 5 forming a fusion oncogene of NPM nucleolar phosphoprotein gene on chromosome 5q35 to tyrosine kinase gene ALK on chromosome 2p23 [5]. Patients with systemic ALK+ ALCL are often younger and have a better prognosis than patients with systemic ALK− ALCL. The origin of angioimmunoblastic T cell lymphoma is the follicular helper T cell and commonly expresses characteristic follicular antigens CXCL13, BCL6, CD10, PD1, ICOS, SAP, and CCR5. In addition, there are recurrent mutations leading to epigenetic dysregulation, such as abnormalities of TET2, IDH2, DNMT3A, RHOA, CD28 and gene fusions of ITL-SYK or CTLA4-CD28 [6,7,8]. The most common type of PTCL in the USA is PTCL-NOS and is a diagnosis of exclusion of other TCL types. The origin of PTCL-NOS is thought to be effector T helper cells, including helper T cells type 1, helper T cells type 2, follicular helper T cells (TFH), and central memory T cells (TCM) which can be established by immunohistochemical analysis. The pathology of PTCL-NOS however does not specifically fit into any other subtype [9]. Adult T cell leukemia/lymphoma is associated with infection with HTLV-1 virus which drives malignant transformation of post-thymic helper T cells that exhibit expression of CD4+CD25+ and upregulation of the regulatory protein FOXp3 [10,11,12,13]. Given the lack of efficacy with traditional chemotherapy for the treatment of PTCL, targeted therapies have been developed with varying success, likely due to the heterogeneous pathophysiology. In this review, we highlight recent developments of novel agents utilized in combination as potential strategies to treat patients with peripheral T cell lymphomas [14].
Current Treatment Paradigms for PTCL
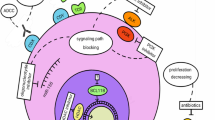
Recently, the prospective COMPLETE study was published revealing common treatment patterns for 273 patients with newly diagnosed PTCL in the USA [15]. Over 45% of patients were 65 years or older and 46% were found to have stage IV disease. Most had PTCL-NOS (51.3%), AITL (26.0%), ALCL (22.7%) with more ALK− (79%) than ALK+ (21%). Of all the patients, 41.8% were started with doxorubicin without etoposide containing regimens and 20.9% with doxorubicin- and etoposide-containing treatments. Response rates were better with doxorubicin-containing regiments with complete response rate of 62.9% compared to 46.4% in non-doxorubicin-based regimens, especially in patients with ALCL. Median follow-up was 26 months (14.4–37.7 months) with median survival of 43 months (95% CI 34.5 months to not reached). The 12- and 24-month survival rates were 71.7 and 58.7%, respectively. Inevitably, disease recurs in most patients and second line treatment options yield limited results. A recent retrospective study conducted at the BCCA showed that median time to relapse or progression after primary therapy was 6.7 months with median survival and progression-free survival after relapse to be 5.5 and 3.1 months, respectively [16•]. The frequencies of histologic subtypes studied were consistent with historic data: the majority was PTCL-NOS (52%), followed by AITL (25%), and then ALCL (23.5%) with more ALK− (66.7%) than ALK+ (30.6%). Those who received chemotherapy after relapse had modest improvement compared to the whole cohort with median OS of 6.5 months and PFS of 3.7 months. While these findings highlight the deficiencies of conventional chemotherapy for the treatment of PTCL, there has been a shift toward targeting identified drivers of disease pathogenesis to reverse or halt T cell lymphomagenesis. Given the broad biological landscape of PTCL, where subtypes often harbor more than one dysregulated pathway, it is likely that simultaneous targeting of multiple pathways will be more efficacious than targeting a singular lesion (Fig. 1).

Landscape of therapeutic targets in T cell lymphoma. Aurora A kinase promotes progression to metaphase by activating CENP-E, a kinesin which transports attached kinetochores to the spindle equator. Alisertib and romidepsin interrupt this process respectively by inhibiting Aurora A kinase and disrupting histone phosphorylation and the attachment of centrosome-associated proteins to the kinetochore. The PI3K/AKT/mTOR pathway is a canonical proliferation pathway that can be inhibited by PI3K inhibitors such as duvelisib, or mTOR inhibitors such as everolimus, and TAK228. CD30 is a member of the TNF receptor family that increases the presence of free NF-kB. NF-kB and IKZF3 (aiolos) are transcription factors that increase the expression of IRF4 (MUM1) and can increase cell proliferation through the effects of Myc. These pathways are targeted by brentuximab, an anti-cd30 agent, as well as lenalidomide, an agent which is able to enhance the proteasomal degradation of IKZF3. Proteasome inhibitors, such as bortezomib and carfilzomib, can inhibit the degradation of many proteins, several which are involved in proapotic mitochondrial pathways. HDAC inhibitors increase the acetylated state of histones, thereby increasing the expression of many tumor suppressors. Inhibitors of DHFR, i.e., pralatrexate, and DNMT, i.e., azacitidine, prevent the silencing of many genes, some of which are active tumor suppressors. (RTK, receptor tyrosine kinase; AAK, Aurora A kinase; CENP-E, centromere-associated protein E; IKK, IκB kinase; IkB, nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRF4, Interferon regulatory factor 4 also known as MUM1; IKZF3, zinc finger protein aiolos also known as ikaros family zinc finger protein 3; DHFR, dihydrofolate reductase; SAM, S-adenosylmethionine; DNMT, DNA methyltransferase; SAH, S-adenosylhomocysteine)
Combination Therapy with Histone Deacetylase Inhibitors
Given in part to the recent understanding that T cell lymphomagenesis is at least partially due to epigenetic dysfunction, especially with mutations identified in TET2, DNMT3A, and IDH2, histone deacetylase (HDAC) inhibitors have demonstrated efficacy in this disease [17,18,19,20]. Most of the novel combination therapies described below include HDAC inhibitors as the backbone of therapy. Four HDAC inhibitors are approved for use around the world for the treatment of T cell lymphomas. They share a common side effect profile of reversible thrombocytopenia, leucopenia, fatigue, and gastrointestinal disturbances. Romidepsin, as monotherapy in relapsed and refractory PTCL, has been shown to have an overall response rate of 25–38% and median duration of response of 8.9–17 months in responders as demonstrated across two phase II trials leading to FDA approval in 2011 [21, 22]. Belinostat was approved following the phase I BELIEF study that treated 31 relapsed/refractory PTCL patients leading to an ORR of 25.8% and median DOR 13.6 months (95% CI 4.5–29.4) [23]. Chidamide (CS055), approved in China, is in the benzamine class of histone deacetylase inhibitor designed to block the catalytic pocket of class I HDACs to inhibit the activity of HDAC1, 2, 3, and 10 leading to growth arrest and apoptosis [24]. In a phase II study of relapsed/refractory PTCL, 79 patients underwent monotherapy with chidamide [25]. Results were notable for ORR of 29%, median DOR 9.9 months, median PFS 2.1 months, and median OS 21.4 months (range 0.3–50.1 months). Vorinostat is a pan-HDAC inhibitor that has been shown to cause growth arrest and caspase-dependent apoptotic and caspase-independent autophagic cell death, with ORR of 29.7% in a phase IIB study of 74 patients with refractory CTCL [26]. This led to FDA approval of vorinostat in treatment of CTCL in 2006. Vorinostat is not approved for PTCL.
Panobinostat is a deacetylase inhibitor that is approved in combination for the treatment of multiple myeloma. It has been shown to mediate genes regulating apoptosis, immune regulation, and angiogenesis in cutaneous T cell lymphoma [27]. Preclinical data in myeloma suggest antineoplastic synergy when bortezomib and panobinostat are combined [28]. A phase III study testing the combination in multiple myeloma shows improved median PFS of 11.99 months (95% CI 10.33–12.94) compared to 8.08 months (CI 95% 7.56–9.23) with bortezomib and placebo leading to benefit in overall survival [29]. This led to FDA approval of panobinostat in multiple myeloma in 2015. Given the applicability of targeting HDACs and the proteasome, this approach has also been evaluated in studies of PTCL (Table 1).
Romidepsin and Pralatrexate
The novel anti-folate drug pralatrexate as a single agent has demonstrated an overall response rate of 29% with median PFS of 3.5 months (95% CI 1.7–4.8) and OS of 14.5 months (95% CI 10.6–22.5) in the phase II PROPEL study [39]. Duration of response was 10.1 months (95% CI 3.4 months—not estimable). Given that pralatrexate and romidepsin were the first two drugs approved for relapsed, refractory PTCL, and have T cell lineage specificity, there is strong rationale for studying the combination. Jain, et al. studied the two drugs in the preclinical setting and demonstrated strong synergy in mouse models of T cell lymphoma [40]. These findings were translated into a phase I study, for patients with relapsed/refractory lymphoma of all subtypes [30•]. Most patients enrolled had T cell lymphoma (N = 14). The overall response rate in the entire population was 47%. Among the T cell lymphoma patients, the overall response rate was 71% (10/14), and 4 of 14 (29%) achieved a complete response. In the T cell lymphoma population, the median PFS was 4.4 months (95% CI 1.2-NA) and median overall survival was 12.4 months (95% CI 8.1-NA). Adverse events were mostly grade 1-2, such as nausea, fatigue, anorexia, diarrhea, and fever. Grade 3-4 toxicities included neutropenia, anemia, thrombocytopenia, mucositis, and sepsis. These toxicities were not seen at the recommended phase 2 dose of pralatrexate 25 mg/m2 and romidepsin 12 mg/m2 given on an every-other-week basis. The results of this study suggest that dual therapy with pralatrexate and romidepsin is an effective and safe platform for patients with PTCL, and is now currently being investigated in a multicenter phase II trial focusing on PTCL (NCT01947140).
Romidepsin and DNA Methyltransferase Inhibitors (DNMT) (Azacitadine, Decitabine)
An epigenetic drug combination that has shown promise in PTCL incorporates romidepsin and DNA methyltransferase (DNMT) inhibitors. This combination could in theory reverse the epigenetic derangements observed in PTCL by simultaneously targeting two epigenetic operations. In a study of patients with relapsed/refractory PTCL, azacitadine monotherapy was shown to have ORR 53% in 19 patients. Five of five patients (100%) of those who achieved a CR were patients with AITL harboring TET2 mutations [41]. Side effects include polyneuropathy and diarrhea. Marchi et al. evaluated the combination of dual epigenetic targeting in preclinical models of PTCL. The authors demonstrated that there was marked synergy when combining HDAC and DNMT inhibitors, such as romidepsin and decitabine. This combination led to increased number of modulated genes from 138 when treated with romidepsin alone, to 390 genes with many of the genes involved in apoptosis and cell cycle arrest [42]. Given these findings, a phase I/IIa clinical trial evaluating oral azacitidine in combination with romidepsin for relapsed refractory lymphoma (NCT01998035) was developed at Columbia University. A total of 27 patients have been enrolled in the phase 1 portion of the study, five of which have TCL [33]. Of the five patients with TCL, four responded to treatment; two of which had a complete response. Among all patients and dose cohorts, there were two grade IV episodes of neutropenia or thrombocytopenia and three grade III toxicities, three of which occurred at the highest doses of azacitidine with romidepsin. Based on these results, a phase II trial is actively enrolling patients with relapsed/refractory PTCL.
Romidepsin and Alisertib
Aurora kinases are a family of serine-threonine kinases that are expressed during mitosis, affecting cell signaling and mitotic division. When expressed in high levels, it is associated with centrosome amplification, mitotic abnormalities, and chromosomal instability, leading to malignancy. One study showed overexpression of aurora kinase was seen in 68% of T cell lymphoma cases [43]. Alisertib (MLN8237), an Aurora A kinase inhibitor, has been shown to have concentration and time-dependent cytotoxic effects on BCL and TCL cell lines [44]. In a phase II study of alisertib monotherapy for patients with relapse/refractory TCL, patients showed an ORR of 30%, median PFS of 3 months (95% CI 2.2–4.3 months), and median DOR of 3 months (range 1–18 months) [45]. The most common side effect was myelosuppression. In a phase III study, alisertib was compared to physician’s choice in relapsed/refractory PTCL, but there was no improved efficacy compared to physician’s choice with ORR of 33% in patients treated with alisertib as monotherapy compared to 40% with pralatrexate, 59% with romidepsin, and 35% with gemcitabine [46]. Due to these data, enrollment was stopped though patients benefiting on the drug were able to continue [47]. In an effort to improve upon these effects, the combination of alisertib plus romidepsin was studied by Zullo et al. and induced more polyploidy and apoptosis, thought to be due to the HDAC inhibitor inducing degradation of AAK as shown in preclinical models of PTCL [48]. The combination of romidepsin and alisertib is currently being studied in patients with relapsed/refractory B or T cell lymphoma (NCT01897012). Preliminary data on 19 patients with relapsed/refractory PTCL (16%), Hodgkin lymphoma (16%), or aggressive BCL (68%) were treated and found to have an ORR of 26% and median PFS 1 month [32]. Of the three patients with PTCL, one patient had a CR, one had stable disease, and one progressed. Eighteen (95%) have discontinued treatment due to progression (78%), completion of treatment (12%), indication for SCT (95%), and patient’s choice (5%). The most common grade 3-4 side effects include myelosuppression, infection, and fatigue.
HDAC Inhibitors Plus Proteasome Inhibitors
Panobinostat is a deacetylase inhibitor that has been shown to mediate genes regulating apoptosis, immune regulation, and angiogenesis in cutaneous T cell lymphoma [27]. A phase Ia/II study on MTD of panobinostat showed that of patients with NHL, one person out of two with T cell lymphoma had a partial response [49]. Proteasome inhibitors such as bortezomib inhibits nuclear factor kappa B, which can be upregulated in some peripheral T cell lymphomas. In a study evaluating T cell lymphoma, monotherapy bortezomib resulted in a response rate of 67% in 12 patients [50]. Fifty percent of those who responded had relapse or progression within 12 months. When tested in T cell lymphoma in an open labeled phase II study with 25 patients, 43% had objective responses (95% CI 23–63) with median duration of response 5.6 months (range 2–33, IQR 1.25–27.5) [38]. Median PFS and OS were 2.59 months (95% CI 0.01–5.42) and 9.90 months (95% CI 3.52–16.26), respectively. Adverse events included diarrhea, peripheral neuropathy, fatigue, vomiting, and cytopenias.
Holkova et al. performed a phase I study of carfilzomib plus vorinostat in patients with relapsed/refractory B cell lymphomas in which most people had progressive disease with only a 5% response rate [51], but given the different underlying biology of B and T cell lymphomas, it is difficult to extrapolate to PTCL patients. To determine effects in relapsed/refractory PTCL, romidepsin plus carfilzomib are being studied in a phase I/II trial (NCT03141203). Interim results have not yet been reported for this study.
Romidepsin and PI3K Inhibitors
Duvelisib (IPI-145) is a phosphoinositide-3-kinase-delta/gamma (PI3K-δ/γ) inhibitor that was used to treat patients with advanced hematologic malignancies in a phase I trial, which included relapsed/refractory indolent NHL, CLL, and TCL patients. Sixteen patients had PTCL and 15 were evaluable. The ORR was 47% in PTCL patients with median overall survival of 36.4 weeks [52]. Grade 3 or higher side effects were seen in 79% of patients with the most common being increased transaminases (36%), rash (21%), and neutropenia (15%). Preclinical data show that duvelisib killed three of four TCL lines that show phosphorylation of AKT compared to zero of seven TCL lines lacking phosphorylation of AKT [31]. Using phosphoproteomic analysis, resistance through PI3Kα activation was overcome by pan-PI3K inhibition or epigenetic reprogramming with the addition of romidepsin. There is a phase I study treating relapsed/refractory T cell lymphomas with duvelisib in combination with romidepsin or bortezomib to determine maximum tolerated dose (NCT02783625). Twelve patients were treated with romidepsin and duvelisib, and eight were evaluable for efficacy. Overall response rate was 50% and median time to response was 51 days (rage 49–54 days). The maximum tolerated dose was defined as duvelisib 75 mg BID and romidepsin 10 mg/m2 on days 1, 8, 15 on a 28-day cycle. The combination was well tolerated with unexpectedly low liver toxicity and manageable cytopenias. The phase II study is now enrolling. For the bortezomib plus duvelisib arm however, there were many toxicities observed such as infections, colitis, and liver toxicity in addition to cytopenias. Of the 17 patients treated, 15 were evaluable for response in which the ORR was 53% and the complete response rate with 20% [53].
HDAC Inhibitors and Lenalidomide
Lenalidomide is an immunomodulatory drug known to target cereblon, an E3 ubiquitin ligase, resulting in antiproliferative and antineoplastic activity across multiple hematologic malignancies, that provided a basis for studying this drug in patients with T cell lymphoma [54]. Phase II studies of lenalidomide in heavily pretreated patients with PTCL led to an ORR of 22–26%, median PFS of 2.5–4 months, median OS of 12 months, and median duration of response of 3.6–13 months [55, 56]. Subgroup analysis revealed AITL patients and those on earlier lines of therapy were the ones who benefitted the most. In AITL patients, lenalidomide monotherapy resulted in ORR 42% (95% CI 23–63%), median PFS was 3.8 months (95% CI 1.9 months-NE), median OS 20.3 months (95% CI 9.1 months-NE), and median duration of response was NE (95% CI 0.5 months-NE) with mean DOR of 5.2 months (range 0–16.6 months) [57]. Side effects included thrombocytopenia, neutropenia, and gastrointestinal symptoms. A phase I/II trial of lenalidomide with vorinostat and dexamethasone in relapsed/refractory PTCL revealed ORR 25%, median PFS 2.2 months and median 6.7 months [35]. Preclinical data combining lenalidomide and romidepsin have shown to induce reactive oxygen species (ROS) in 53–61.8% of cells in TCL cell-line models compared to 17–23% with romidepsin alone or 11% with lenalidomide alone, suggesting this combination induces cytotoxicity through mitochondria-mediated apoptosis [58]. A phase I study of combination romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma revealed activity in T cell lymphoma [36]. Ten patients had relapsed/refractory PTCL and achieved an ORR of 50% (5/10 PR) with median time to response of 7.3 weeks (range 2.8–16.9 weeks). Grade 3 or higher adverse events were seen in 71%, mostly due to neutropenia (48%), thrombocytopenia (38%), anemia (33%), and electrolyte derangements (43%). Given the longer median PFS, OS, and DOR in monotherapy lenalidomide studies of previously untreated patients, improvement on the combination of targeting this biology was sought. A phase II study in untreated PTCL with lenalidomide and romidepsin is ongoing (NCT02232516).
Romidepsin was also studied in combination with lenalidomide and carfilzomib in a three drug regimen. Romidepsin and carfilzomib were given on days 1 and 8 and lenalidomide was given on days 1–14 of a 21-day cycle. Twenty-four patients were evaluable for MTD determination which was defined as romidepsin 8 mg/m2, lenalidomide 15 mg, and carfilzomib 36 mg/m2. The dose-limiting toxicity was thrombocytopenia. Of the 16 patients evaluable for efficacy with PTCL, the ORR was 50% with a CR rate of 31%. The median event-free survival was 13.6 weeks. A phase II study is now accruing patients [37].
Chidamide is also being studied in combination with prednisone, cyclophosphamide, and thalidomide in relapsed or refractory peripheral T cell lymphoma [34]. Twelve Chinese patients were studied with ORR 83.3%, MTR 3 months, and OR along with PFS not reached. Adverse effects were neutropenia, thrombocytopenia, anemia, and EBV reactivation.
Combination Therapy with Brentuximab Vedotin
Brentuximab vedotin is an anti-CD30 monoclonal antibody conjugated by a protease-cleavable linker to antimicrotubule agent monomethylauristatin E that has demonstrated activity against systemic ALCL (sALCL), leading to its approval for this indication in the relapsed setting. Fifty-eight patients with relapsed/refractory CD30+ sALCL were treated with monotherapy brentuximab in a phase II study and were found to have an ORR of 86% (95% CI 74.6–93.9%) with 57% CR, median DOR 12.6 months (95% CI 5.7 months-NE), and median PFS 13.3 months (95% CI 6.9 months-NE) [59]. Five-year follow-up revealed 79% OS and 57% PFS among those with CR [60]. Adverse effects included neuropathy, nausea, fatigue, pyrexia, diarrhea, rash, constipation, and neutropenia with one patient with TLS. When applied to relapsed/refractory CD30+ AITL (n = 13) and PTCL-NOS (n = 22), brentuximab led to an ORR of 54% in AITL and 33% in PTCL-NOS [61]. Median DOR was 5.5 and 7.6 months with median PFS 6.7 and 1.6 months in AITL and PTCL-NOS, respectively.
Brentuximab Combination Studies
Several studies have recently been activated for the study of brentuximab in combination with other agents known to have activity in PTCL. These studies are very early on and to date do not have any reported interim results. A study is assessing the feasibility of romidepsin and brentuximab in cutaneous T cell lymphoma (NCT02616965).
The combination of brentuximab vedotin and lenalidomide has been studied in 18 patients with relapsed/refractory DLBCL with treatment-related adverse events including anemia (50%), thrombocytopenia (33%), neutropenia (28%), elevated ALT (28%), hypokalemia (28%), hypocalcemia (22%), and peripheral neuropathy (22%) [62]. This combination will also be used to treat patients with CD30+ relapsed/refractory PTCL, CTCL, or Hodgkin lymphoma starting December 15, 2017 (NCT03302728).
The mammalian target of rapamycin (mTOR) operates in two multiprotein complexes, complex 1 and complex 2 (TORC1, TORC2), as it regulates cell growth in the PI3K/AKT/mTOR pathway. TAK228 (formerly MLN0128) is an oral dual TORC1/2 inhibitor that has been studied in patients with relapsed/refractory multiple myeloma (n = 26), non-Hodgkin lymphoma (n = 3), or Waldenstrom’s macroglobinemia (n = 4). The NHL subtypes were DLBCL and mantle cell lymphoma. One patient with NHL discontinued prior to first assessment and the remaining two had stable disease [63]. Adverse effects included nausea, fatigue, hyperglycemia, and myelosuppression. To study the merits of targeting CD30 and MTOR, a phase I study of brentuximab vedotin and TAK228 is ongoing to study MTD in patients with relapsed/refractory classical Hodgkin lymphoma, ALCL, and other CD30+ PTCL (NCT03205891).
Combinations of Novel Agents with Chemotherapy
Chemotherapy is a standard first choice in therapy and generally leads to responses that may successfully bridge patients to stem cell transplant for consolidation. However, as previously mentioned, relapse often occurs and overall survival dramatically decreases after first line therapy fails. Many of the studies combining chemotherapy with novel agents that have been published thus far demonstrate greater toxicity with only limited added benefit (Table 2).
Chemotherapy with Romidepsin
Romidepsin was combined with CHOP in a phase IB/II study in previously untreated PTCL patients. Of 35 evaluable patients, the ORR was 68% with an OS and PFS at 18 months of 76.5 and 57%, respectively, though the author warned that longer follow-up time was needed [68]. Common serious adverse events included febrile neutropenia (13.5%), general health deterioration (13.5%), lung infection (10.8%), and vomiting (8%). The phase III study is ongoing (NCT01796002).
Chemotherapy with Lenalidomide
Combination CHOEP with lenalidomide was studied as a phase I trial in patients with newly diagnosed PTCL. CHOEP was administered at standard doses with escalating levels of lenalidomide with doses higher than 10 mg displayed significant hematologic toxicities. Non-hematologic side effects include diarrhea, hypotension, and mucositis [64]. At last update of trial results, three patients have proceeded to ASCT and three have proceeded to lenalidomide maintenance. The phase II cohort is currently accruing to evaluate response rates (NCT02561273). There is also an ongoing study evaluating the efficacy of treatment in older patients with CHOP chemotherapy plus lenalidomide specifically in newly diagnosed angioimmunoblastic T cell lymphoma (NCT01553786). Interim analysis showed ORR of 54% of 37 patients who were evaluable out of the 38 patients enrolled [65]. Grade 4 neutropenia and thrombocytopenia were seen in 70 and 30% of patients, respectively. Four episodes of thrombosis occurred during treatment without any secondary malignancies reported.
Chemotherapy with Pralatrexate
In a phase II study using COEP and pralatrexate, 34 patients with newly diagnosed PTLC were enrolled [66]. The ORR was 70% with 2-year OS and PFS of 60% (95% CI 39–76) and 39%, respectively, with improvement in rates if patients received HDT/SCT. Adverse effects included myelosuppression, elevated creatinine, and liver transaminases. There was not sufficient added benefit to warrant further study, but given improvement in outcomes with anthracycline-based chemotherapy, there currently is a dose optimizing trial for pralatrexate and CHOP in PTCL (NCT02594267). Interim results from this study indicate that the MTD was not reached therefore the recommended phase 2 dose was pralatrexate 30 mg/m2 on days 1 and 8 along with CHOP chemotherapy on a 21-day cycle. Of the 31 patients enrolled on the phase 1 and expansion cohort, 14 patients experienced mucositis and 5 patients had grade ≥ 3 neutropenia. Of the 27 patients evaluated for response, the ORR and CR rates were 89 and 67%, respectively [70].
Chemotherapy and Chidamide
Chidamide is now being studied with chemotherapy regimens CHOEP, CHOP, and ICE in China in newly diagnosed and relapsed/refractory patients with PTCL (NCT02987244, NCT02809573, NCT03268889, NCT02856997).
Chemotherapy and Everolimus
Everolimus acts on the mTOR pathway. In vitro data suggests that everolimus inhibits the phosphorylation of mTOR at autophosphorylation sites and reduces levels of c-Myc in TCL [71]. However, everolimus-treated cells developed increased phosphorylation at certain sites suggesting feedback mechanisms protecting cells from apoptosis. A phase II study demonstrated that everolimus with CHOP led to an ORR of 90% though limited by relapse with the 2-year PFS of 33% and median PFS of 11.2 months [67]. The authors postulated that the high relapse rate may be secondary to the feedback mechanisms seen in vitro. The 2-year OS was 70%. Side effects included hematologic toxicity, mucositis, GI symptoms, peripheral neuropathy, and infections. Given the response rate, this regimen could be used as a bridge to ASCT if administered quickly. The regimen theoretically could be improved upon if feedback mechanisms leading to resistance can be prevented.
Chemotherapy and Brentuximab
In the phase I trial of ECHELON-2 with 39 patients, combination brentuximab and CHP were compared to sequential brentuximab followed by CHOP in the upfront treatment of CD30+ mature T cell lymphomas [72]. Thirteen were randomized to the sequential group, and 26 patients were in the combination group. Brentuximab in combination with CHP led to 100% ORR with 88% CR, 1-year OS of 88% (95% CI 68–96%), and PFS of 71% (95% CI 11.7-NE) compared to the sequential arm which resulted in a 100% ORR with 1-year OS 85% (95% CI 51–96%) and 1-year PFS 77% (95% CI, 44–92%). Two patients experienced POD while receiving sequential CHOP. Adverse effects were similar as prior studies with the addition of more GI side effects when chemotherapy was added. Three-year follow-up of combination treatment led to OS 80% (95% CI 59, 91 months), median PFS not reached (95% CI 12.3, not reached) [69]. Phase III of ECHELON-2 is ongoing (NCT01777152). Other similar studies include NCT03217643, NCT01309789, and NCT03113500.
There are also studies focusing on specific subtypes of PTCL. Brentuximab vedotin with methotrexate, L-asparaginase, and dexamethasone (MAD) is being studied in an effort to treat newly diagnosed extranodal NK/T cell lymphoma in a phase I/II study. (NCT03246750). ATLL patients who were previously untreated or failed a maximum of one therapy are being studied in a phase II trial of BV-CHEP (NCT03264131).
Immunotherapy
Data on immunotherapy and its efficacy in PTCL is in its infancy, but promising data is emerging. Several PTCL subtypes are associated with the Epstein Bar virus (EBV) which has been demonstrated to modulate PD-1 and PD-L1 on the tumor and microenvironment. EBV is a DNA virus that infects B cells and expresses latency genes that drive transformation and proliferation. There are four patterns of gene expression, termed Latency 0, 1, 2, and 3. Latency 3 is most immunogenic, but as EBV gets recognized and cleared, infected B cells downregulate expression of immunogenic proteins leading to persistence. This same biology leads to tumor evasion from immune surveillance and potential drug resistance [73]. These principles of viral latency may also be applicable to viruses such as HTLV-1 and others.
In a phase I study of Nivolumab, an anti-programmed-death-1 antibody, five patients were enrolled with PTCL of whom two had a response with a PFS of 14 weeks [74]. Pembrolizumab, another anti-programmed-death-1 antibody, was used for treating seven patients with relapsed/refractory NK/T cell lymphoma failing L-asparaginase with 100% ORR after 6-month follow-up, leading to second line approval [75]. This was used off-label but considering EBV as a known oncogene of NK/T cell lymphoma, it is a viral antigen that is an ideal target for immunotherapy. An international phase Ib study of pembrolizumab with decitabine and pralatrexate is pending recruitment (NCT03240211).
Durvalumab, a human monoclonal antibody that blocks the interaction of PD-L1 with PD-1 and CD80, is also being studied in different combinations with pralarexate, romidepsin, and 5-azacitadine in an open label phase 1/2a study for patients with relapsed or refractory PTCL (NCT03161223). In addition, a phase II study using durvalumab and lenalidomide in relapsed/refractory NK/T cell lymphoma is ongoing (NCT03054532).
Conclusion
Peripheral T cell lymphomas are rare diseases but can be devastating to patients and their families given the high relapse rate and limited options in the relapsed/refractory setting. There has been great progress in understanding the mechanisms leading to lymphomagenesis, albeit these are heterogeneous diseases with a multitude of oncogenic drivers making a unified management approach difficult. While developing tailored therapies for patients, we must keep the underlying biology in mind as we consider which combinations will be most successful. PTCL often initially responds to treatment but these diseases are adaptive, inevitably becoming resistant to therapy. Combination therapies targeting biology driving PTCL are promising for the management of these patients because of their potential synergy which could lead to deeper responses, but more work needs to be done to learn about tumor feedback and resistance mechanisms. Many combination studies now allow treatment for patients in the upfront setting due to poor overall results. Hopefully with the growing interest in this disease subtype, robust improvements in outcomes will be realized in the near future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. https://doi.org/10.1182/blood-2016-01-643569. With the new WHO classification, discrete entities of PTCL are now being identified by molecular and phenotypic markers. This new classification is critical to our ability to define disease entities which may respond to certain classes of targeted therapy.
Foss F. T-cell lymphomas. New York: Humana Press; 2012.
Vose J, Armitage J, Weisenburger D, International, T. C. L. P. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–30. https://doi.org/10.1200/JCO.2008.16.4558.
Durkop H, et al. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell. 1992;68(3):421–7. https://doi.org/10.1016/0092-8674(92)90180-K.
Morris SW, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. https://doi.org/10.1126/science.8122112.
Dupuis J, Boye K, Martin N, Copie-Bergman C, Plonquet A, Fabiani B, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper T cells. Am J Surg Pathol. 2006;30(4):490–4. https://doi.org/10.1097/00000478-200604000-00009.
de Leval L, Rickman DS, Thielen C, Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11):4952–63. https://doi.org/10.1182/blood-2006-10-055145.
Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014;4(12):e264. https://doi.org/10.1038/bcj.2014.83.
Matsumoto Y, Horiike S, Ohshiro M, Yamamoto M, Sasaki N, Tsutsumi Y, et al. Expression of master regulators of helper T-cell differentiation in peripheral T-cell lymphoma, not otherwise specified, by immunohistochemical analysis. Am J Clin Pathol. 2010;133(2):281–90. https://doi.org/10.1309/AJCP0SBHYVLY5EML.
Yamada Y. Phenotypic and functional analysis of leukemic cells from 16 patients with adult T-cell leukemia/lymphoma. Blood. 1983;61(1):192–9.
Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br J Haematol. 2004;126(1):81–4. https://doi.org/10.1111/j.1365-2141.2004.04999.x.
Karube K, Aoki R, Sugita Y, Yoshida S, Nomura Y, Shimizu K, et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod Pathol. 2008;21(5):617–25. https://doi.org/10.1038/modpathol.2008.25.
Barbi J, Pardoll DM, Pan F. Ubiquitin-dependent regulation of Foxp3 and Treg function. Immunol Rev. 2015;266(1):27–45. https://doi.org/10.1111/imr.12312.
Lue JK, Kress A, Amengual JE. Therapeutic options for aggressive T-cell lymphomas. Curr Hematol Malig Rep. 2017;12(4):269–81. https://doi.org/10.1007/s11899-017-0389-7.
Carson KR, Horwitz SM, Pinter-Brown LC, Rosen ST, Pro B, Hsi ED, et al. A prospective cohort study of patients with peripheral T-cell lymphoma in the United States. Cancer. 2017;123(7):1174–83. https://doi.org/10.1002/cncr.30416.
• Mak V, Hamm J, Chhanabhai M, Shenkier T, Klasa R, Sehn LH, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31(16):1970–6. https://doi.org/10.1200/JCO.2012.44.7524. Survival for patients with relapsed PTCL is dismal and highlights the need for improved therapies.
Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38. https://doi.org/10.1016/j.ccr.2011.06.003.
Mercher T, Quivoron C, Couronné L, Bastard C, Vainchenker W, Bernard OA. TET2, a tumor suppressor in hematological disorders. Biochim Biophys Acta. 2012;1825(2):173–7. https://doi.org/10.1016/j.bbcan.2011.12.002.
Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–6. https://doi.org/10.1056/NEJMc1111708.
Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–3. https://doi.org/10.1182/blood-2011-11-391748.
Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–34. https://doi.org/10.1182/blood-2010-10-312603.
Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631–6. https://doi.org/10.1200/jco.2011.37.4223.
O’Connor OA, Horwitz S, Masszi T, van Hoof A, Brown P, Doorduijn J, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492–9. https://doi.org/10.1200/jco.2014.59.2782.
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69(4):901–9. https://doi.org/10.1007/s00280-011-1766-x.
Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 2015;26(8):1766–71. https://doi.org/10.1093/annonc/mdv237.
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. https://doi.org/10.1200/JCO.2006.10.2434.
Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14(14):4500–10. https://doi.org/10.1158/1078-0432.CCR-07-4262.
Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95(5):794–803. https://doi.org/10.3324/haematol.2009.015495.
San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–206. https://doi.org/10.1016/s1470-2045(14)70440-1.
• Amengual JE, Lichtenstein R, Lue J, Sawas A, Deng C, Lichtenstein E, Khan K, Atkins L, Rada A, Kim HA, Chiuzan C, Kalac M, Marchi E, Falchi L, Francescone MA, Schwartz L, Cremers S, O’Connor OA. A phase I study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood. 2017;blood-2017-09-806737 https://doi.org/10.1182/blood-2017-09-806737. Combination of the first two approved drugs in PTCL lymphoma is well tolerated and leads to a high rate of response.
Moskowitz AJKR, Mehta-Shah N, Myskowski P, Kheterpal M, Dogan A, Davey T, Galassa N, Evan M, Shah M, Ganesan N, Lubin L, Kim YH, Khodadoust M, Almazan T, Dai J, Jacobsen ED, Weinstock DM, Horwitz SM. In vitro, in vivo, and parallel phase I evidence support the safety and activity of duvelisib, a PI3K-δ,γ inhibitor, in combination with romidepsin or bortezomib in relapsed/refractory T-cell lymphoma. 2017.
Strati POY, Fayad LE, Nastoupil L, Fowler NH, Hagemeister FB, Kwak LW, Lee HJ, Wang, W, Westin JR, Ruben C, Wesson E, Fanale MA. A Phase 1 trial of alisertib and romidepsin for relapsed/refractory aggressive B-cell and T-cell lymphomas. 2017.
O’Connor OAL, J. K, Amengual JE, Sawas A, Deng C, Lichtenstein E, et al. Oral azacytadine (AZA) and romidepsin (R) reveals promising activity in patients with relapsed or refractory (R/R) periphearl T-cell lymphoma (PTCL). Hematol Oncol. 2017;35:274–5. https://doi.org/10.1002/hon.2438_147.
Xu WZ,H, Wang L, Liang J, Kong Y, Wu W, Fan L, et al. The efficacy and tolerance of chidamide, prednisone, cyclophosphamide and thalidomide (C-PCT) in relapsed or refractory peripheral T-cell lymphoma: a pilot study. Hematol Oncol. 2017;35:397. https://doi.org/10.1002/hon.2439_167.
Hopfinger G, Nösslinger T, Lang A, Linkesch W, Melchardt T, Weiss L, et al. Lenalidomide in combination with vorinostat and dexamethasone for the treatment of relapsed/refractory peripheral T cell lymphoma (PTCL): report of a phase I/II trial. Ann Hematol. 2014;93(3):459–62. https://doi.org/10.1007/s00277-014-2009-0.
Mehta-Shah N, et al. A phase I/II trial of the combination of romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma: activity in T-cell lymphoma. J Clin Oncol. 2015;33:8521. https://doi.org/10.1200/jco.2015.33.15_suppl.8521.
Mehta-Shah N, et al. A phase Ib/IIa trial of the combination of romidepsin, lenalidomide and carfilzomib in patients with relapsed/refractory lymphoma shows complete responses in relapsed and refractory B- and T-cell lymphomas. Blood. 2017;130:821.
Tan D, Phipps C, Hwang WY, Tan SY, Yeap CH, Chan YH, et al. Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: an open-label, multicentre phase 2 trial. Lancet Haematol. 2015;2(8):e326–33. https://doi.org/10.1016/S2352-3026(15)00097-6.
O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182–9. https://doi.org/10.1200/JCO.2010.29.9024.
Jain S, Jirau-Serrano X, Zullo KM, Scotto L, Palermo CF, Sastra SA, et al. Preclinical pharmacologic evaluation of pralatrexate and romidepsin confirms potent synergy of the combination in a murine model of human T-cell lymphoma. Clin Cancer Res. 2015;21(9):2096–106. https://doi.org/10.1158/1078-0432.ccr-14-2249.
Delarue R, Dupuis J, Sujobert P, Barbieux S, Marçais A, Tournilhac O, et al. Treatment with hypomethylating agent 5-azacytidine induces sustained response in angioimmunoblastic T cell lymphomas. Blood. 2016;128:4164.
Marchi E, Zullo KM, Amengual JE, Kalac M, Bongero D, McIntosh CM, et al. The combination of hypomethylating agents and histone deacetylase inhibitors produce marked synergy in preclinical models of T-cell lymphoma. Br J Haematol. 2015;171(2):215–26. https://doi.org/10.1111/bjh.13566.
Kanagal-Shamanna R, Lehman NL, O’Donnell JP, Lim MS, Schultz DS, Chitale DA, et al. Differential expression of aurora-A kinase in T-cell lymphomas. Mod Pathol. 2013;26(5):640–7. https://doi.org/10.1038/modpathol.2012.211.
Zullo KM, Guo Y, Cooke L, Jirau-Serrano X, Mangone M, Scotto L, et al. Aurora A kinase inhibition selectively synergizes with histone deacetylase inhibitor through cytokinesis failure in T-cell lymphoma. Clin Cancer Res. 2015;21(18):4097–109. https://doi.org/10.1158/1078-0432.CCR-15-0033.
Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul Haq M, et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T-cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol. 2015;33(21):2399–404. https://doi.org/10.1200/JCO.2014.60.6327.
O’Connor OAOM, Jacobsen ED, Vidal JMR, Trotman J, Demeter J, Masszi T, Prereira J, Ramchandren R, d’AMore FA, Foss F, Kim WS, Leonard JP, Chiattone CS, Zinzani PL, Liu H, Jung JA, Zhou X, et al. First multicenter, randomized phase 3 study in patients (Pts) with relapsed/refractory (R/R) peripheral T-cell lymphoma (PTCL): alisertib (MLN8237) versus investigator’s choice (Lumiere trial; NCT01482962). 2015.
O’Connor OAOM, Jacobsen ED, Vidal JMR, Trotman J, Demeter J, Masszi T, et al. First multicenter, randomized phase 3 study in patients (pts) with relapsed/refractory (R/R) peripheral T-cell lymphoma (PTCL): alisertib (MLN8237) versus investigator’s choice (Lumiere trial; NCT01482962). Blood. 2015;126:341.
Park JH, Jong HS, Kim SG, Jung Y, Lee KW, Lee JH, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating aurora-A kinase. J Mol Med (Berl). 2008;86(1):117–28. https://doi.org/10.1007/s00109-007-0260-8.
DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T, et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013;27(8):1628–36. https://doi.org/10.1038/leu.2013.38.
Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(27):4293–7. https://doi.org/10.1200/JCO.2007.11.4207.
Holkova B, Kmieciak M, Bose P, Yazbeck VY, Barr PM, Tombes MB, et al. Phase 1 trial of carfilzomib (PR-171) in combination with vorinostat (SAHA) in patients with relapsed or refractory B-cell lymphomas. Leuk Lymphoma. 2016;57(3):635–43. https://doi.org/10.3109/10428194.2015.1075019.
Horwitz SP,P, Flinn I, Kahl BS, Sweeney J, Stern HM, Douglas M, et al. Duvelisib (IPI-145), a phosphoinositide-3-kinase-δ,γ inhibitor, shows activity in patients with relapsed/refractory T-cell lymphoma. Blood. 2014;124:803.
Moskowitz AJ, et al. In vitro, in vivo, and parallel phase I evidence support the safety and activity of duvelisib, a PI3K-δ, γ inhibitor, in combination with romidepsin or bortezomib in relapsed/refractory T-cell lymphoma. Blood. 2017;130:819.
Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9. https://doi.org/10.1126/science.1244917.
Toumishey E, Prasad A, Dueck G, Chua N, Finch D, Johnston J, et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer. 2015;121(5):716–23. https://doi.org/10.1002/cncr.29103.
Morschhauser F, Fitoussi O, Haioun C, Thieblemont C, Quach H, Delarue R, et al. A phase 2, multicentre, single-arm, open-label study to evaluate the safety and efficacy of single-agent lenalidomide (Revlimid) in subjects with relapsed or refractory peripheral T-cell non-Hodgkin lymphoma: the EXPECT trial. Eur J Cancer. 2013;49(13):2869–76. https://doi.org/10.1016/j.ejca.2013.04.029.
Ishida T, Fujiwara H, Nosaka K, Taira N, Abe Y, Imaizumi Y, et al. Multicenter phase II study of lenalidomide in relapsed or recurrent adult T-cell leukemia/lymphoma: ATLL-002. J Clin Oncol. 2016;34(34):4086–93. https://doi.org/10.1200/JCO.2016.67.7732.
Cosenza M, et al. The histone deacetylase inhibitor romidepsin synergizes with lenalidomide and enhances tumor cell death in T-cell lymphoma cell lines. Cancer Biol Ther. 2016;1–13. https://doi.org/10.1080/15384047.2016.1219820.
Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–6. https://doi.org/10.1200/JCO.2011.38.0402.
Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood. 2017:blood-2017-05-780049. https://doi.org/10.1182/blood-2017-05-780049.
Horwitz SM, Advani RH, Bartlett NL, Jacobsen ED, Sharman JP, O’Connor OA, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123(20):3095–100. https://doi.org/10.1182/blood-2013-12-542142.
Ward JPT,J, Luo J, Wagner-Johnston ND, Cashen AF, Fehniger TA, Bartlett NL. A phase I trial of brentuximab vedotin in combination with lenalidomide in relapsed or refractory diffuse large B-cell lymphoma. Blood. 2015;126:3988.
Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, et al. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenstrom’s macroglobulinemia. Am J Hematol. 2016;91(4):400–5. https://doi.org/10.1002/ajh.24300.
Lunning MH,S, Mehta-Shah M, Moskowitz A, Advani R, Beaven A, Haverkos B, et al. Phase I/II study of CHOEP plus lenalidomide as initial therapy for patients with stage II-IV peripheral T-cell non-Hodgkin lymphoma: phase I results from the T-cell consortium. Hematol Oncol. 2017;35:411–2. https://doi.org/10.1002/hon.2439_190.
Safar VDLL, Meignan M, Bachy E, Cartron G, Moles-Moreau MP, Delmer A, Bouabdallah R, Voillat L, Corront B, Casasnovas O, Cacheux V, Gressin R, Robreau Y, Pallardy S, Tilly H, Delfau-Larue MH, Gaulard P, Haioun C. Prospective phase II trial of lenalidomide in association with CHOP in elderly patients with angioimmunoblastic T cell lymphoma (AITL): interim analysis of a LYSA study. 2015.
Advani RH, Ansell SM, Lechowicz MJ, Beaven AW, Loberiza F, Carson KR, et al. A phase II study of cyclophosphamide, etoposide, vincristine and prednisone (CEOP) alternating with pralatrexate (P) as front line therapy for patients with peripheral T-cell lymphoma (PTCL): final results from the T- cell consortium trial. Br J Haematol. 2016;172(4):535–44. https://doi.org/10.1111/bjh.13855.
Kim SJ, Shin DY, Kim JS, Yoon DH, Lee WS, Lee H, et al. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Ann Oncol. 2016;27(4):712–8. https://doi.org/10.1093/annonc/mdv624.
Dupuis JM,F, Ghesquieres H, Tilly H, Casasnovas O, Amorim S, Ribrag V, et al. Final analysis of the RO-CHOP phase Ib/II study: romidepsin in association with CHOP in patients with peripheral T-cell lymphoma (PTCL). Blood. 2014;124:504.
Horwitz SSAR, Forero-Torres A, Bartlett NL, Advani R, Pro B, Chen R, Davies AJ, Illidge T, Huebner D, Kennedy DA, Fanale MA. Frontline treatment of CD30+ peripheral T-cell lymphomas with brentuximab vedotin in combination with CHP: 3-year durability and survival follow-up. 2015.
Shustov A, et al. Pralatrexate in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) in previously untreated patients with peripheral T-cell lymphoma (PTCL): a phase 1 dose-escalation study. Blood. 2017;130:818.
Witzig TE, Reeder C, Han JJ, LaPlant B, Stenson M, Tun HW, et al. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma. Blood. 2015;126(3):328–35. https://doi.org/10.1182/blood-2015-02-629543.
Fanale MA, Horwitz SM, Forero-Torres A, Bartlett NL, Advani RH, Pro B, et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol. 2014;32(28):3137–43. https://doi.org/10.1200/JCO.2013.54.2456.
Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59–73. https://doi.org/10.1038/cr.2016.153.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34(23):2698–704. https://doi.org/10.1200/JCO.2015.65.9789.
Kwong YL, Chan TSY, Tan D, Kim SJ, Poon LM, Mow B, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood. 2017;129(17):2437–42. https://doi.org/10.1182/blood-2016-12-756841.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Helen Ma and Ardy Davarifar each declare no potential conflicts of interest.
Jennifer E. Amengual is a section editor for Current Hematologic Malignancy Reports.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on T-Cell and Other Lymphoproliferative Malignancies
Rights and permissions
About this article

Cite this article
Ma, H., Davarifar, A. & Amengual, J.E. The Future of Combination Therapies for Peripheral T Cell Lymphoma (PTCL). Curr Hematol Malig Rep 13, 13–24 (2018). https://doi.org/10.1007/s11899-018-0432-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-018-0432-3