
Abstract
Purpose of Review
Central sleep apnea occurs in up to 50% of heart failure patients and worsens outcomes. Established therapies are limited by minimal supporting evidence, poor patient adherence, and potentially adverse cardiovascular effects. However, transvenous phrenic nerve stimulation, by contracting the diaphragm, restores normal breathing throughout sleep and has been shown to be safe and effective. This review discusses the mechanisms, screening, diagnosis, and therapeutic approaches to CSA in patients with HF.
Recent Findings
In a prospective, multicenter randomized Pivotal Trial (NCT01816776) of transvenous phrenic nerve stimulation with the remedē System, significantly more treated patients had a ≥ 50% reduction in apnea-hypopnea index compared with controls, with a 41 percentage point difference between group difference at 6 months (p < 0.0001). All hierarchically tested sleep, quality of life, and daytime sleepiness endpoints were significantly improved in treated patients. Freedom from serious related adverse events at 12 months was 91%. Benefits are sustained to 36 months.
Summary
Transvenous phrenic nerve stimulation improves quality of life in patients with heart failure and central sleep apnea. Controlled trials evaluating the impact of this therapy on mortality/heart failure hospitalizations and “real world” experience are needed to confirm safety and effectiveness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Central sleep apnea (CSA) is a sleep breathing disorder distinguished by a temporary interruption of neural output from the brain’s respiratory control center resulting in loss of respiratory stimulation and airflow cessation [1, 2]. Polysomnogram (PSG) studies have reported a high prevalence of CSA (20–50%) in ambulatory patients with stable heart failure (HF) [3]. Like obstructive sleep apnea (OSA), CSA occurs primarily during sleep. However, in patients with HF, one form of CSA, periodic breathing, or Cheyne-Stokes respiration, can also occur during wakefulness and it is a marker of advanced disease. This review discusses the mechanisms, screening, diagnosis, and therapeutic approaches to CSA in patients with HF.
Mechanisms of Central Sleep Apnea in Heart Failure
In HF, various components of the negative feedback system controlling breathing are altered during both sleep and wakefulness. Furthermore, specific sleep-related events justify the predominance of CSA during sleep. Prolonged arterial circulation time delays arrival of information on changes in Po2 and Pco2 from pulmonary capillary blood to the central chemoreceptors. This, together with negative feedback loop’s propensity toward instability in response to a ventilatory disturbance and decreased functional residual capacity, heightens the probability of the periodic breathing typical of the CSA occurring in HF patients [4•]. A short pause in breathing normally causes compensatory hyperventilation. If this response results in a carbon dioxide level lower than that needed to restore normal breathing, the system becomes unstable and will oscillate between underventilation and overventilation, possibly converting a negative into a positive feedback system.
Another causative factor for CSA is increased sensitivity of chemoreceptors to CO2 so that when Pco2 rises or the Po2 drops, the inappropriate hyperventilation drives the Pco2 below the apneic threshold, resulting in breathing cessation. Consequently, a rise in Pco2 and a fall in Po2 initiate and perpetuate the cycles of hyperventilation and hypoventilation [4•]. The variability in chemosensitivity may explain why CSA is not ubiquitous in HF patients. In HF, for a given episode of hyper- or hypoventilation, changes in Po2 and Pco2 will be exaggerated [4•, 5•]. This results in a marked compensatory ventilatory response, which further destabilizes breathing. In HF patients, the already reduced functional residual capacity, due to excess of intrathoracic fluid, cardiomegaly, and stiffness of the respiratory system, can further drop in the supine position, increasing the likelihood of periodic breathing. Importantly, additional reductions in metabolic rate and cardiac output during sleep facilitate the occurrence of CSA in HF patients.
Like OSA, CSA can also occur in the daytime when a patient is awake. During sleep, CSA originates from the withdrawal of the non-chemical drive of wakefulness on breathing and the Pco2 level being below the apneic threshold when rhythmic breathing stops [4•, 5•]. The difference between the prevailing Pco2 minus the Pco2 at the apneic threshold (Pco2 reserve) is essential to the development of CSA and the smaller the difference, the greater is the probability of apnea events. In healthy individuals, with the onset of sleep, ventilation decreases and Pco2 increases. If the prevailing Pco2 exceeds the apneic threshold, rhythmic breathing continues. In contrast, in some HF patients, the prevailing Pco2 does not sufficiently rise with onset of sleep, and CO2 chemosensitivity also increases compared with that of normal breathing [4•, 5•]. Thus, the smaller difference between prevailing and apneic threshold Pco2 augments the probability of CSA events during sleep. The increase of chemosensitivity above that of normal breathing is especially detrimental during the arousals that follow apneas, when hyperventilation lowers the prevailing Pco2 closer to the apneic threshold [4•, 5•]. Although it is unclear why some HF patients lack a normal increase in Pco2, a potential causative factor is the absence of normal sleep-induced decrease in ventilation. Furthermore, increased venous return in the supine position may augment pulmonary capillary pressure which, in turn, may result in the hyperventilation that prevents the expected rise in Pco2 during sleep. In addition, higher pulmonary capillary pressure increases chemosensitivity below normal breathing and decreases the Pco2 reserve, further enabling CSA. Also, vagal afferents modulate responses of both carotid bodies and central chemoreceptors [4•, 5•]. Notably, in HF patients, the predictive value of a low awake arterial Pco2 (< 35 mmHg) for the development of CSA during sleep is approximately 80% [4•, 5•]. However, a low awake arterial Pco2 is not a prerequisite for CSA. Even in HF patients with a normal awake arterial Pco2, the key predictors of CSA are the narrow difference between prevailing arterial and apneic threshold Pco2 and increased CO2 chemosensitivity compared with that of normal breathing.
Like OSA, CSA is associated with increased sympathetic activity which can be improved by effective continuous positive airway pressure (CPAP) therapy and oxygen [6]. Most studies show an association of CSA with decreased survival in HFrEF patients [7,8,9,10,11,12]. Comparison of 56 CSA with 32 non-CSA HF patients followed for 4.2 years showed that after controlling for 24 variables, CSA was independently associated with increased mortality (HR = 2.14; p = 0.02) [13]. In HF patients, CSA is also an independent predictor of re-hospitalization. In a prospective observational study of consecutive patients with an average LVEF of 22%, 165 CSA subjects had a 1.5 times higher rate ratio (p = 0.03) for cardiac readmissions at 1 and 6 months compared with 139 patients without sleep apnea after adjustment for age, gender, body weight, blood pressure, coronary artery disease, hemoglobin, serum sodium and creatinine concentration, diabetes mellitus, and length of stay [14]. Worsening of HF likely results from the repeated episodes of apnea, hypoxia, reoxygenation, and arousal which occur throughout the night in patients with CSA. In the long term, these pathologic effects lead to further sustained sympathetic nervous system activation, oxidative stress, systemic inflammation, and endothelial dysfunction [5•].
Diagnosis and Screening
Clinical
The diagnosis of CSA in HF patients is difficult because the symptoms of this sleep disorder and HF often overlap [15••, 16]. This overlap of HF and CSA symptoms contributes to the underdiagnosis of sleep-related breathing disorders in HF patients. Symptoms shared by CSA and HF include characteristics of sleep onset and maintenance insomnia, nocturia, orthopnea, paroxysmal nocturnal dyspnea and hyperpnea during periodic breathing, perception of not feeling refreshed upon awakening, and daytime fatigue. With CSA, there are additional diagnostic challenges in HF patients lacking the hallmarks of OSA, such as obesity and snoring [4•, 5•]. In addition, screening questionnaires for OSA have not been validated in CSA. Diagnosis of CSA in HF patients requires a high index of clinical suspicion and attention to important clues, such as high NYHA functional class, frequent HF hospitalizations,, low steady-state arterial Pco2, atrial fibrillation (AF), nocturnal ventricular arrhythmias, and the complaint of daytime fatigue rather than sleepiness [4•, 5•]. Furthermore, although CSA is more likely to occur at low LVEF, it can also occur in 25–35% of HF patients with preserved LVEF [4•, 5•].
Sleep Studies
In-Laboratory Attended Polysomnogram
The gold standard for diagnosing sleep disorders requires an in-laboratory PSG attended by a technician where oxygen saturation, oronasal airflow, respiratory movement, electroencephalogram, body position, electromyogram, electrooculogram, and electrocardiogram are recorded. Although PSG provides detailed and highly accurate results, it requires an overnight stay, a sleep technician, and manual scoring of the data. This process is expensive, and results are often delayed. Furthermore, elderly patients may lack transportation to the PSG laboratory [4•, 5•]. These shortcomings have spurred the development of alternative diagnostic devices that allow sleep evaluation in the patient’s home [16].
Home Sleep Apnea Testing
A description of all commercially available devices for home detection of sleep disorders is beyond the scope of this article. Some common characteristics include use of fewer sensors than with an attended PSG, greater patients’ comfort, and use of automatic algorithms running within the device or on commercial computer software. Importantly, because these devices do not measure the components of sleep, they can underestimate the severity of disease. However, these devices can serve as good screening tools to identify sleep disordered breathing as a basis for referral to a Sleep Physician. Most home sleep apnea testing (HSAT) utilize a chest or abdominal belt, oxygen sensor, and airflow. However, one newer device (WatchPAT, Itamar Medical, Atlanta, GA) utilizes a proprietary peripheral arterial tone signal (PAT) and records heart rate, oximetry, actigraphy, body position, snoring, and chest motion using three points of contact (chest sensor, wrist bracelet, and finger probe). The raw data is immediately downloaded and autoscored. The PAT signal is a non-invasive measure of the arterial pulsatile volume changes at the fingertip. Attenuation of the PAT signal and acceleration of pulse rate indicate sympathetic activation which reflects autonomic arousals occurring during sleep disordered breathing. Combined with oximetry desaturations and re-saturations, a proprietary algorithm calculates apnea/hypopnea index (AHI), respiratory disturbance index, and oxygen desaturation index (ODI). Importantly, the device has a module for the specific identification of CSA. The technology also includes a HIPAA-compliant cloud-based IT process for secure data transfer and interpretation as well as a cloud-based platform for long-term monitoring of therapy delivery [17•, 18,19,20,21].
Treatment
Pharmacological
Optimization of Guideline-Directed Medical Therapy
The cornerstone of CSA treatment in HF patients is meticulous application of guideline-directed medical therapy (GDMT) [22, 23]. Effective reduction of pulmonary congestion can increase Pco2 reserve [24]. Furthermore, GDMT, by decreasing arterial circulation time with improvement in fluid excess and stroke volume, can improve functional residual capacity and contribute to stabilize breathing. Beta blockers are essential because these drugs blunt the nocturnal cardiac sympathetic hyperactivity produced by recurrent arousals and desaturations. Among the three beta blockers recommended by HF guidelines, carvedilol may be preferable for HF patients with CSA because it does not inhibit melatonin which may improve sleep and is secreted through the cyclic adenosine monophosphate-mediated beta-adrenergic signal transduction pathway [25]. Cardiac resynchronization therapy is also associated with CSA improvement [26,27,28,29,30]. In patients with Stage D HF, heart transplantation, but not mechanical circulatory support, is associated with resolution of CSA [31, 32]. Often, however, optimal GDMT does not achieve adequate suppression of CSA.
Other Pharmacologic Approaches
Nocturnal Oxygen
Supplemental nocturnal oxygen may improve CSA by increasing the difference between prevailing and apneic threshold Pco2, decreasing ventilatory response to CO2, and improving pulmonary oxygen content [33]. Several small studies with modest follow-up support improvement of CSA by nocturnal oxygen supplementation [4•]. However, prospective placebo-controlled long-term studies are needed to determine if this therapy also decreases mortality in HFrEF patients. The ongoing Impact of Low Flow Nocturnal Oxygen Therapy on Hospital Admissions and Mortality in Patients with Heart Failure and Central Sleep Apnea (LOFT-HF) trial (NCT03745898) will hopefully answer this question upon its projected completion in February 2023.
Inhaled CO2 and Addition of External Dead Space
A few studies have shown that low-level CO2 inhalation and increased Pco2 with additional external dead space may improve CSA. In contrast, other studies caution that this therapy may worsen arousals, promoting neuro-hormonal activation. Thus, use of CO2 and external dead space to treat CSA should be avoided in HF patients [34,35,36,37].
Theophylline
This drug stimulates respiration by competing for receptor sites with adenosine, a respiratory depressant in the central nervous system. However, theophylline does not increase ventilatory response to CO2. Enthusiasm about the observed decrease in AHI and improved arterial oxyhemoglobin saturation by theophylline demonstrated in very small studies is lessened by the arrhythmogenic potential of phosphodiesterase inhibition [33, 34].
Acetazolamide
Acetazolamide improves CSA by increasing functional residual capacity, acting as a mild diuretic and improving loop diuretic-induced alkalemia. In a small double-blind placebo-controlled crossover study of 12 HF patients, acetazolamide significantly reduced central AHI and arterial oxyhemoglobin desaturation, and improved subjective experience of overall sleep quality, feeling rested upon awakening, unintentional daytime dozing, and fatigue. However, therapy with acetazolamide can be associated with paresthesias, tinnitus, nausea, vomiting, diarrhea, and drowsiness [38].
Benzodiazepines
The potential role of these drugs in CSA is the decrease in arousals. However, benzodiazepines are ineffective in HF patients with CSA in whom they may increase risk of OSA events [39].
Non-pharmacologic Treatment
Positive Airway Pressure Devices
Continuous Positive Airway Pressure
Several devices, including CPAP, bilevel pressure, and autoservo-ventilation (ASV), have been used to treat CSA in HF patients, but response to therapy is inconsistent. In the Canadian Continuous Positive Airway Pressure for Central Sleep Apnea and Heart Failure (CANPAP) trial that randomized 132 patients to CPAP and 128 to usual care, 47% of treatment subjects were non-responsive to CPAP at 3 months [40]. Compared with the control arm, CPAP-treated patients experienced a 50% decrease in AHI, significantly less oxygen desaturation, lower average plasma norepinephrine levels, and higher LVEF. However, CPAP-treated patients had lower transplantation-free survival, which lead to premature discontinuation of the study. Importantly, the average nightly duration of CPAP therapy at 1 year was 3.6 h. A post hoc analysis that compared outcomes of patients with suppression of CSA with those of non-responders revealed that responders had significantly improved transplant-free survival [41]. Because most deaths in non-responders were due to HF progression or sudden cardiac death, it has been hypothesized that these subjects’ biventricular function was preload dependent: any reduction in venous return by increased intrathoracic pressure from CPAP could reduce right ventricular stroke volume and return to the left ventricle, decreasing LV stroke volume and causing hypotension, diminished coronary blood flow, myocardial ischemia, and arrhythmias. These effects are magnified during sleep when, normally, blood pressure decreases [4• 5•].
Adaptive Servo-ventilation
Until the publication of the results of the Treatment of Predominant Central Sleep Apnea by Adaptive Servo Ventilation in Patients With Heart Failure (SERVE-HF) trial, this technology was recommended for CPAP intolerant and/or unresponsive CSA patients [42]. Devices for ASV augment ventilation only when the patient’s minute ventilation decreases below a given target, thereby eliminating periodic breathing [42]. New generations of ASV devices also include automatic end expiratory positive pressure algorithms to treat mixed CSA and OSA [42]. This feature could be helpful during HF decompensation, when excess fluid shifts from the lower extremities to the neck in the supine position, causing upper airway obstruction [42,43,44,45,46,47,48,49,50,51]. Pre-SERVE-HF studies showed that in HF patients with CSA, ASV was superior to CPAP or bilevel PAP in terms of AHI reduction, improvement of HF biomarkers and LVEF, and decrease in combined endpoints of mortality and HF hospitalizations [42,43,44,45,46,47,48,49,50,51]. However, SERVE-HF, a large multicenter international clinical trial that studied the effects of treating CSA with an ASV device in subjects with advanced HFrEF, has shown that ASV is unsafe as it causes an excess cardiovascular mortality [52]. Although the investigators hypothesized that CSA might be a compensatory mechanism with a protective effect in HFrEF, there are more plausible explanations for SERVE-HF’s unfavorable outcomes. These include small amounts of residual CSA associated with profound oxygen desaturation, poor treatment adherence, high crossover rates, and the use of an obsolete ASV device which delivered higher minute ventilation and pressures than expected. Based on the results of SERVE-HF, all ASV devices are currently contraindicated for HFrEF patients with CSA. The Effect of Adaptive Servo Ventilation (ASV) on Survival and Hospital Admissions in Heart Failure (ADVENT-HF) plans to recruit 860 HFrEF subjects with either OSA or CSA that will be randomized to either GDMT alone or GDMT plus ASV with a new-generation device to determine the effects of ASV on death and HF hospitalizations (NCT01128816).
Transvenous Phrenic Nerve Stimulation
Rationale
A novel physiological therapy which induces normal breathing by transvenously stimulating the phrenic nerve has been developed and evaluated in clinical trials. The remedē System (Respicardia, Minnetonka, Minnesota) is a totally implantable lead-based device that delivers unilateral transvenous phrenic nerve stimulation (TPNS) to cause diaphragmatic contraction in a manner that reproduces normal breathing [53]. Diaphragm contraction creates a negative intrathoracic pressure like that generated by normal breathing, so that airflow is augmented and CSA episodes during sleep are decreased. Suppression of impending central apneas prevents the increases in Pco2 and decreases in Po2 that trigger hyperventilation and reduction in Pco2 below the apneic threshold [53].
The remedē System Device
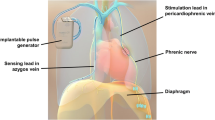
The remedē System consists of a neurostimulator (similar in appearance to a standard pacemaker), a stimulation lead, and a sensing lead [53]. The neurostimulator is placed in either the left or right pectoral region, with a preference for the right side to accommodate existing or future CIEDs. Depending on individual patient venous anatomy, the stimulation lead is advanced into either the left pericardiophrenic or right brachiocephalic vein, to stimulate a nerve. Lead placement is verified by fluoroscopic confirmation of movement of the diaphragm with stimulation. The sensing lead is typically placed in the azygos vein to sense respiration by thoracic impedance [54] (Fig. 1, panel a). The system is designed to automatically stimulate the phrenic nerve during the scheduled time at night when the patient is asleep and in a reclining position, which is detected by a position and motion sensor present in the device. The system is typically activated 1 month after implant, according to an algorithm that applies a stimulation pattern that enables full diaphragmatic contraction while the patient continues to sleep. The ranges of pulse stimulation are typically 0.1–10.0 mA for 60–300 μs at 10–40 Hz [53]. Over approximately 12 weeks, stimulation is automatically programmed to gradually increase until diaphragmatic capture is consistently achieved without disrupting sleep [53].

Panel a The remedē System. Panel b Percentage change in AHI at 6 months’ follow-up compared with baseline for patients in the treatment group and control group who have 6-month polysomnography results. The remaining seven (10%) patients from the treatment group without polysomnography data who were included in the intention-to-treat analysis for primary effectiveness were imputed as failures. Panel c Percentage of patient responses to the patient global assessment. Patients were asked, “Specifically in reference to your overall health, how do you feel today as compared to how you felt before having your device implanted: markedly improved, moderately improved, mildly improved, no change, slightly worse, moderately worse, or markedly worse?”. Panel d Median AHI, ArI, CAI, and ODI4 are displayed by visit for subjects in the treatment group. A PSG was performed at baseline, 6, 12, 18, and 24 months. A home sleep study was performed at 36 months (33 subjects had not reached this visit at time of study closure). AHI, apnea/hypopnea index; ArI, arousal index; CAI, central apnea index; Mo, month. ODI4, oxygen desaturation ≥ 4% index. Reproduced, with permission, from refs. 55,56,57, and
Pilot Study of Transvenous Phrenic Nerve Stimulation
A prospective multicenter non-randomized pilot study of chronic TPNS with the remedē System in CSA patients showed that from baseline to 3 months, chronic TPNS improved the AHI by 55% (p < 0.001), central apnea index (CAI) by 83% (p < 0.001), 4% oxygen desaturation index (ODI4) by 52% (p < 0.001), arousal index by 35% (p < 0.001), rapid eye movement (REM) sleep by 41% (p < 0.001), patient global assessment (PGA) by 45%, and Minnesota Living With Heart Failure Questionnaire score (MLWHFQ) by an average of 10 points (p < 0.001) [58]. Efficacy was maintained at 6 and 12 months and therapy was well tolerated. Serious adverse events related to the device, implantation procedure, or therapy were consistent with those occurring with other CIEDs at a similar stage of development [59, 60].
Pivotal Trial of Transvenous Phrenic Nerve Stimulation
The encouraging results of this pilot study led to the launch of the prospective multicenter, randomized remedē System Pivotal Trial at 31 sites in Germany, Poland, and the USA (NCT01816776) [55]. Subjects were required to have been medically stable for at least 30 days and have received appropriate GDMT, be aged ≥ 18 years, be expected to tolerate study procedures, and willing and able to comply with study requirements. Potential subjects prospectively underwent a qualifying attended PSG within 40 days before implant. Subjects were enrolled if they had CSA meeting the following criteria based on a PSG scored by a central core laboratory: AHI ≥ 20 events per hour of sleep, central apneas ≥ 50% of all apneas, ≥ 30 central apnea events throughout the night, and an OAI ≤ 20% of the total AHI. Eligible patients (most with comorbidities including 64% with HF and only a small percentage with idiopathic CSA) underwent device implantation and were randomly assigned to either treatment (active TPNS therapy) or control (no stimulation for 6 months). The control group had TPNS therapy activated after the 6-month assessments. In the intention-to-treat population, the primary effectiveness endpoint was the comparison of the proportions of patients in the treatment versus control groups achieving ≥ 50% AHI reduction from baseline to 6 months, measured by an attended PSG assessed in a core laboratory by sleep specialists blinded to randomized treatment assignment. The primary safety endpoint of 12-month freedom from serious adverse events related to the procedure, system, or delivered therapy was evaluated in all patients. Of 151 eligible patients, 73 were assigned to TPNS and 78 to control. The intention-to-treat analysis showed that significantly more patients in the TPNS group than in the control group had a ≥ 50% AHI reduction at 6 months (51% vs. 11%), for a difference between groups of 41% (95% CI 25–54; p < 0.0001). Additionally, all hierarchically tested secondary endpoints pertaining to sleep variables and quality of life were significantly better in TPNS than in controls at 6 months (Table 1 and Fig. 1, panel b). Average implant time was 2.7 ± 0.8 h, with a success rate of 97% and lead revision rate of 3.4%. Freedom from serious adverse events related to the implant procedure, the remedē System, or the delivered therapy at 12 months was 91% (95% CI 85–97%). None of seven deaths occurring within the 12 months was related to implant, system, or therapy. Of 73 TPNS patients, 27 (37%) reported non-serious therapy-related discomfort that was resolved with system reprogramming in all but one subject. In 64 (42%) patients with CIEDs, no ventricular arrhythmias were adjudicated as caused by neurostimulation. In one ICD recipient, inappropriate defibrillation was triggered by oversensing, which was corrected by remedē System reprogramming without reoccurrences [55].
Based on these results, the remedē System received approval for clinical use by the Food and Drug Administration on October 6, 2017.
The remedē System Pivotal Trial had some limitations. Despite recruitment efforts, mostly whites and a low percentage of women met the eligibility criteria. A mitigating factor is the documented predominance of all sleep disorders, and particularly CSA, in males. Subjective patient assessments of health status (PGA and ESS) may have been biased by knowing the treatment assignment. However, the risk of bias is lessened with the scoring of objective sleep measures by masked core laboratory investigators. In addition, a patient experience questionnaire was completed by both groups after 6 months of active therapy at the request of the Food and Drug Administration. Patients were asked, “Based on your experience with the remedē System therapy, would you elect to have this medical device implanted again?” In the intent-to-treat treatment group with a 6-month visit, 58/62 patients (94%, 95% CI 84–98%) responded affirmatively. In the intent-to-treat former control group with a 12-month visit (6 months of active therapy), 66/68 patients (97%, 95% CI 90–100%) also responded positively. Daytime central apneas occur in CSA patients [4•, 5•]. Although improvement in sleep quality by nighttime TPNS may mitigate the harmful effects of daytime central apneas, the effects of nighttime TPNS on daytime CSA have not been evaluated in the study [55].
Despite these limitations, the achievement of both primary safety and effectiveness endpoints and of all hierarchically tested secondary endpoints in the remedē System Pivotal Trial confirms that TPNS is a promising therapy for CSA of different etiologies. The consistency between favorable changes in sleep variables and quality of life measures demonstrates that the therapy produces clinically meaningful improvements (Fig.1, panel c). Importantly, delivery of the therapy occurs throughout the night and is independent of patients’ adherence, one of the key limitations of mask-based therapies. In previous studies of PAP devices, only 60% of patients had adequate therapy adherence, defined as an average use of 4 h per night [41, 61, 62]. Since TPNS works automatically and continuously throughout the night, it is not affected by patient adherence and can potentially reduce the overall apnea burden for all hours of sleep. Use of TPNS was associated with improved nocturnal oxygenation, an important observation considering the findings by Oldenburg and colleagues that in patients with sleep disordered breathing time with oxygen saturation below 90% is an independent predictor of all-cause mortality [63]. In contrast with other therapies which reduce AHI but not arousals, TNPS was associated with a significant decrease in arousals, which are associated with sympathetic activation. The improvement in the arousal index and REM sleep accompanying the reduction in AHI with TPNS suggest amelioration of sleep quality, which may explain the improvement of PGA and Epworth Sleepiness Score (ESS), further supporting the clinical relevance of the effects of TPNS [55].
Importantly, additional analyses of the remedē System Pivotal Trial conducted in both treatment and former control (in whom TPNS was activated at 6 months) demonstrated reproducibility of the treatment effect in the former control group and enduring effectiveness and safety up to 36 months [56, 57, 64] (Fig.1, panel d). A detailed description of long-term device therapy has been recently published. In addition, monitoring of long-term safety continues in the remedē System Post Approval Study (NCT03425188).
Per-Protocol Analysis of Transvenous Phrenic Nerve Stimulation in Heart Failure Patients
The fact that CSA predicts a poor prognosis in HF patients stimulated a per-protocol analysis of the 96 (64%) remedē System Pivotal Trial HF subjects to determine if TPNS is associated with changes in HF-specific metrics. In this HF cohort, in addition to improvement in sleep metrics, MLHFQ scores changed by − 6.8 ± 20.0 points from baseline after 12 months of therapy (p = 0.005). At 6 months, there was evidence of a potential signal for a reduced HF hospitalization rate in the treatment compared with the control group (4.7% vs. 17.0%; p = 0.065). In HF patients with a LVEF ≤ 45% and no permanent atrial fibrillation (n = 50), results suggested a possible reduction in left ventricular end-systolic volume at 12 months (median change − 6.0 mL; p = 0.078) accompanied by an increase in the median LVEF of 4.0% (p = 0.004). The 12-month safety results were similar to that of the overall population [65].
Taken together, the analyses summarized above support the notion that TPNS is a promising therapy for the treatment of CSA. Key additional investigations are needed, including mechanistic studies of the hemodynamic effects of negative intrathoracic pressure in HF patients, impact of TPNS on daytime CSA, registry databases to evaluate real world experience with TPNS, and, most importantly, outcomes trials evaluating the effects of TPNS on mortality and HF hospitalizations in a large population with longer term follow-up [66,67,68,69]. In the interim, the ongoing post-approval study will contribute to answer questions on 5-year durability of TPNS’ effectiveness, safety, and device battery longevity. As of the time of this writing, 60 sites have the remedē System available for clinical implantation.
Conclusions
The presence of CSA is associated with poor outcomes, even after adjustment for the appropriate prognostic markers. Therefore, attention to clinical clues and a low threshold for performing screening tests is appropriate. Given the debilitating symptoms and poor outcomes, treatment of CSA is a clinically meaningful therapeutic target. The effectiveness of pharmacological therapies is doubtful and PAP therapies are limited by patient tolerability, acceptance, adherence, and safety concerns. The novel approach of TPNS causes diaphragmatic contraction, creating negative intrathoracic pressure like normal breathing. The remedē System Pivotal Trial, which enrolled patients with moderate-severe CSA and serious comorbidities, including 64% of patients with underlying HF, confirmed that TPNS is safe and effective in patients with CSA due to multiple etiologies. The safety and effectiveness of TPNS is durable, as confirmed by subjects’ follow-up through 36 months of active therapy. Beside improvements in sleep indices, sleep architecture, daytime sleepiness, and quality of life, the remedē System Pivotal Trial was the first randomized study to show improvements in arousals and REM sleep in CSA patients. Furthermore, TPNS works automatically and continuously throughout the night for every hour of sleep and does not depend on patient adherence, the Achilles’ heel of mask-based therapies. In HFrEF patients, TPNS was associated with a signal for improvement in cardiac remodeling and reduction in HF hospitalizations. Randomized controlled trials evaluating the impact of TPNS on mortality and HF hospitalizations, as well as “real world” clinical experiences with TPNS, are needed to further elucidate the role of TPNS in the treatment of CSA.
Abbreviations
- AF:
-
Atrial fibrillation
- AHI:
-
Apnea/hypopnea index
- ASV:
-
Adaptive servo-ventilation
- CAI:
-
Central apnea index
- CIED:
-
Cardiac implantable electronic device
- CPAP:
-
Continuous positive airway pressure
- CSA:
-
Central sleep apnea
- GDMT:
-
Guideline-directed medical therapy
- HF:
-
Heart failure
- HIPAA:
-
Health insurance portability and accountability act
- LVEF:
-
Left ventricular ejection fraction
- MLWHFQ:
-
Minnesota Living With Heart Failure Questionnaire
- ODI:
-
Oxygen desaturation index
- OSA:
-
Obstructive sleep apnea
- PAT:
-
Peripheral arterial tone
- PGA:
-
Patient global assessment
- PSG:
-
Polysomnogram
- REM:
-
Rapid eye movement
- TPNS:
-
Transvenous phrenic nerve stimulation
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: pathophysiology and treatment. Chest. 2007;131:595–607.
Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3:141–63.
Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, et al. Sleep apnea in 81 ambulatory male patients with stable heart failure: types and their prevalences, consequences, and presentations. Circulation. 1998;97:2154–9.
• Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69:841–58 Superb and comprehensive review of the links between sleep disordered breathing and cardiovascular disease.
• Costanzo MR, Khayat R, Ponikowski P, et al. Mechanisms and clinical consequences of untreated central sleep apnea in heart failure. J Am Coll Cardiol. 2015;65:72–84 Comprehensive analysis of the pathophysiological consequences of central sleep apnea, including neuro-hormonal activation, oxidative stress, and inflammation.
Sin DD, Logan AG, Fitzgerald FS, Liu PP, Bradley TD. Effects of continuous positive airway pressure on cardiovascular outcomes in heart failure patients with and without Cheyne-Stokes respiration. Circulation. 2000;102:61–6.
Jilek C, Krenn M, Sabeh D, et al. Prognostic impact of sleep disordered breathing and its treatment in heart failure: an observational study. Eur J Heart Fail. 2011;13:68–75.
Hanly P, Zuberi-Khokhar N. Increased mortality associated with Cheyne-Stokes respiration in patients with congestive heart failure. Am J Respir Crit Care Med. 1996;153:272–6.
Leite JJ, Mansur AJ, de Freitas HFG, Chizola PR, Bocchi EA, Terra-Filho M, et al. Periodic breathing during incremental exercise predicts mortality in patients with chronic heart failure evaluated for cardiac transplantation. J Am Coll Cardiol. 2003;41:2175–81.
Brack T, Thuer I, Clarenbach CF, et al. Daytime Cheyne-Stokes respiration in ambulatory patients with severe congestive heart failure is associated with increased mortality. Chest. 2007;132:1463–71.
Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol. 2007;49:2028–34.
Silva RS, Figueiredo AC, Mady C, Lorenzi-Filho G. Breathing disorders in congestive heart failure: gender, etiology and mortality. Braz J Med Biol Res. 2008;41:215–22.
Yoshihisa A, Shimizu T, Owada T, Nakamura Y, Iwaya S, Yamauchi H, et al. Adaptive servo ventilation improves cardiac dysfunction and prognosis in chronic heart failure patients with Cheyne-Stokes respiration. Int Heart J. 2011;52:218–23.
Khayat R, Abraham W, Patt B, Brinkman V, Wannemacher J, Porter K, et al. Central sleep apnea is a predictor of cardiac readmission in hospitalized patients with systolic heart failure. J Card Fail. 2012;18:534–40.
•• Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373:1095–105 Main results of the SERVE-HF trial, in which adaptive servo-ventilation improved sleep measures but was associated with increased cardiovascular mortality in patients with central sleep apnea and advanced heart failure.
Mendonca F, Mostafa SS, Ravelo-Garcia AG, Morgado Dias F, Penzel T. Devices for home detection of obstructive sleep apnea: a review. Sleep Med Rev. 2018;41:149–60.
• Bar A, Pillar G, Dvir I, Sheffy J, Schnall R, Lavie P. Evaluation of a portable device based on peripheral arterial tone for unattended home sleep studies. Chest. 2003;123(3):695e703 Informative analysis of home sleep studies devices. Home sleep studies are preferred by patients and are less expensive than in center polysomnograms.
Pillar G, Bar A, Betito M, Schnall R, Dvir I, Sheffy J, et al. An automatic ambulatory device for detection of AASM defined arousals from sleep: the WP100. Sleep Med. 2003;4(3):207e12.
Pittman S, Pittman N, MacDonald M, Malhotra A, Fogel R, White D. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(5):923e33.
Zou D, Grote L, Peker Y, Lindblad U, Hedner J. Validation a portable monitoring device for sleep apnea diagnosis in a population-based cohort using synchronized home polysomnography. Sleep. 2006;29(3):367e74.
Pang K, Gourin C, Gourin T. A comparison of polysomnography and the WatchPAT in the diagnosis of obstructive sleep apnea. Otolaryngol Head Neck Surg. 2007;137(4):665e8.
Ponikowski P, Voors AA, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–200.
Yancy CW, Jessup M, Bozkurt B, et al. ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2013;62:e147–239.
Chenuel B, Smith C, Skatrud J, et al. Increased propensity of apnea in response to acute elevations in left atrial pressure during sleep in the dog. J Appl Physiol. 2006;101:76–83.
Stoschizky K, Sakotnik A, Lercher P, et al. Influence of beta-blockers on melatonin release. Eur J Clin Pharmacol. 1999;55:111–5.
Sinha AM, Skobel EC, Breithardt OA, Norra C, Markus KU, Breuer C, et al. Cardiac resynchronization therapy improves central sleep apnea and Cheyne-Stokes respiration in patients with chronic heart failure. JACC. 2004;44:68–71.
Gabor JY, Newman DA, Barnard-Roberts V, Korley V, Mangat I, Dorian P, et al. Improvement in Cheyne-Stokes respiration following cardiac resynchronization therapy. Eur Respir J. 2005;26:95–100.
Oldenburg O, Faber L, Vogt J, Dorszewski A, Szabados F, Horstkotte D, et al. Influence of cardiac resynchronization therapy on different types of sleep disordered breathing. Eur J Heart Fail. 2007;9:820–6.
Kara T, Novak M, Nykodym J, Bybee KA, Meluzin J, Orban M, et al. Short-term effects of cardiac resynchronization therapy on sleep-disordered breathing in patients with systolic heart failure. Chest. 2008;134:87–93.
Stanchina ML, Ellison K, Malhotra A, Anderson M, Kirk M, Benser ME, et al. The impact of cardiac resynchronization therapy on obstructive sleep apnea in heart failure patients. Chest. 2007;132:433–9.
Javaheri S, Abraham W, Brown C, et al. Prevalence of obstructive sleep apnea and periodic limb movement in 45 subjects with heart transplantation. Eur Heart J. 2004;25:260–6.
Lee Goldberg Case report.
Sasayama S, Izumi T, Matsuzaki M, Matsumori A, Asanoi H, Momomura SI, et al. Improvement of quality of life with nocturnal oxygen therapy in heart failure patients with central sleep apnea. Circ J. 2009;73:1255–62.
Khayat RN, Xie A, Patel AK, Kaminski A, Skatrud JB. Cardiorespiratory effects of added dead space in patients with heart failure and central sleep apnea. Chest. 2003;123:1551–60.
Szollosi I, Jones M, Morrel MJ, et al. Effect of CO2 inhalation on central sleep apnea and arousals from sleep. Respiration. 2004;71:493–8.
Lorenzo-Filho G, Rankin F, Bies I, et al. Effects of inhaled carbon dioxide and oxygen on Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 1999;159:1490–8.
Mebrate Y, Willson K, Manisty CH, Baruah R, Mayet J, Hughes AD, et al. Dynamic CO2 therapy in periodic breathing: a modeling study to determine optimal timing and dosage regimes. J Appl Physiol. 2009;107:696–706.
Javaheri S, Sands SA, Edwards BA. Acetazolamide attenuates Hunter-Cheyne-Stokes breathing yet paradoxically augments the hypercapnic ventilatory response in patients with heart failure. Ann Am Thorac Soc. 2014;11:80–6.
Biberdorf DJ, Steens R, Millar TW, Kryger MH. Benzodiazepines in congestive heart failure: effects of temazepam on arousability and Cheyne-Stokes respiration. Sleep. 1993;16:529–38.
Bradley T, Logan A, Kimoff J, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2006;353:2025–33.
Arzt M, Floras JS, Logan AG, Kimoff RJ, Series F, Morrison D, et al. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: a post hoc analysis of the Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure Trial (CANPAP). Circulation. 2007;115:3173–80.
Javaheri S, Goetting MG, Khayat R, et al. The performance of two automatic servo-ventilation devices in the treatment of central sleep apnea. Sleep. 2011;34:1693–8.
Teschler H, Döhring J, Wang YM, et al. Adaptive pressure support servo-ventilation. Am J Respir Crit Care Med. 2001;64:614–9.
Sharma BK, Bakker JP, McSharry DG, et al. Adaptive servo-ventilation for treatment of sleep-disordered breathing in heart failure: a systematic review and meta-analysis. Chest. 2012;142:1211–21.
Pepperell J, Maskell NA, Jones DR, et al. A randomized controlled trial of adaptive ventilation for Cheyne-Stokes breathing in heart failure. Am J Respir Crit Care Med. 2003;168:1109–14.
Hetland A, Haugaa KH, Olseng M, Gjesdal O, Ross S, Saberniak J, et al. Three-month treatment with adaptive servo-ventilation improves cardiac function and physical activity in patients with chronic heart failure and Cheyne-Stokes respiration: a prospective randomized controlled trial. Cardiology. 2013;126:81–96.
Arzt M, Schroll S, Series F, et al. Auto-servo-ventilation in heart failure with sleep apnoea: a randomized controlled trial. Eur Respir J. 2013;42:1244–54.
Kasai T, Kasagi S, Maeno K-I, et al. Adaptive servo-ventilation in cardiac function and neurohormonal status in patients with heart failure and central sleep apnea nonresponsive to continuous positive airway pressure. JACC Heart Fail. 2013;1:58–63.
Kasai T, Usui Y, Yoshioka T, et al. Effect of flow-triggered adaptive servo-ventilation compared with continuous positive airway pressure in patients with chronic heart failure with coexisting obstructive sleep apnea and Cheyne-Stokes respiration. Circ Heart Fail. 2010;3:140–8.
Randerath WJ, Nothofer G, Priegnitz C, et al. Long-term auto-servo-ventilation or constant positive pressure in heart failure and coexisting central with obstructive sleep apnea. Chest. 2012;142:440–7.
Galetke W, Ghassemi BM, Priegnitz C, Stieglitz S, Anduleit N, Richter K, et al. Anticyclic modulated ventilation versus continuous positive airway pressure in patients with coexisting obstructive sleep apnea and Cheyne–Stokes respiration: a randomized crossover trial. Sleep Med. 2014;15:874–9.
Cowie MR, Wegscheider K, Teschler H. Adaptive servo-ventilation for central sleep apnea in heart failure. N Engl J Med. 2016;374:690–1.
Costanzo MR, Augostini R, Goldberg LR, Ponikowski P, Stellbrink C, Javaheri S. Design of the remedē System Pivotal Trial: a prospective, randomized study in the use of respiratory rhythm management to treat central sleep apnea. J Card Fail. 2015;21:892–902.
Augostini RS, Afzal MR, Costanzo MR, et al. How to implant a phrenic nerve stimulator for treatment of central sleep apnea? J Cardiovasc Electrophysiol. 2019;30:792–9.
• Costanzo MR, Ponikowski P, Javaheri S, Augostini R, Goldberg L, Holcomb R, et al. remede System Pivotal Trial Study Group. Transvenous neurostimulation for central sleep apnoea: a randomised controlled trial. Lancet. 2016;388:974–82 Main results of the pivotal trial of transvenous phrenic nerve stimulation.
Fox H, Oldenburg O, Javaheri S, et al. Long-term efficacy and safety of phrenic nerve stimulation for the treatment of central sleep apnea. SLEEPJ. 2019;42:1–9.
Fudim M, Spector AR, Costanzo MR, Pokorney SD, Mentz RJ, Jagielski D, et al. Phrenic nerve stimulation for the treatment of central sleep apnea: a pooled cohort analysis. J Clin Sleep Med. 2019;15:1747–55.
Abraham WT, Jagielski D, Oldenburg O, Augostini R, Krueger S, Kolodziej A, et al. Phrenic nerve stimulation for the treatment of central sleep apnea. JCHF. 2015;3:360e9.
Alonso C, Leclercq C, d’Allonnes FR, Pavin D, Victor F, Mabo P, et al. Six year experience of transvenous left ventricular lead implantation for permanent biventricular pacing in patients with advanced heart failure: technical aspects. Heart. 2001;86:405e10.
Jagielski D, Ponikowski P, Augostini R, Kolodziej A, Khayat R, Abraham WT. Transvenous stimulation of the phrenic nerve for the treatment of central sleep apnoea: 12 months’ experience with the Remedē ® System. Eur J Heart Fail. 2016;18:1386–93.
Takama N, Kurabayashi M. Effect of adaptive servo-ventilation on 1-year prognosis in heart failure patients. Circ J. 2012;76:661–7.
Hetzenecker A, Escourrou P, Kuna ST, Series F, Lewis K, Birner C, et al. Treatment of sleep apnea in chronic heart failure patients with auto-servo ventilation improves sleep fragmentation: a randomized controlled trial. Sleep Med. 2016;17:25–31.
Oldenburg O, Wellmann B, Buchholz A, Bitter T, Fox H, Thiem U, et al. Nocturnal hypoxaemia is associated with increased mortality in stable heart failure patients. Eur Heart J. 2016;37:1695–703.
Costanzo MR, Ponikowski P, Javaheri S, et al. Sustained 12 month benefit of phrenic nerve stimulation for central sleep apnea. Am J Cardiol. 2018;121:1400–8.
Costanzo MR, Ponikowski P, Coats A, Javaheri S, Augostini R, Goldberg LR, et al. Phrenic nerve stimulation to treat patients with central sleep apnoea and heart failure. Eur J Heart Fail. 2018;20:1746–54.
Gutleben KJ, Fox H, Sommer P, Rudolph V, Nölker G. Interventional techniques to increase implantation success of transvenous phrenic nerve stimulation for central sleep apnea treatment. Sleep Breath. 2019. https://doi.org/10.1007/s11325-019-01917-0.
Arzt M. Neurostimulation in patients with heart failure and central sleep apnoea. Lancet. 2016;388:938–40.
Valika A, Costanzo MR. Sleep-disordered breathing during congestive heart failure: to intervene or not to intervene? Card Fail Rev. 2017 Nov;3(2):134–9.
Abraham WT, Pleister A, Germany R. Identification and treatment of central sleep apnoea: beyond SERVE-HF Card. Fail Rev. 2018;4:50–3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Costanzo is a Consultant for Respicardia; Principal Investigator, remedē System Pivotal Trial.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of Topical Collection on Devices
Rights and permissions
About this article

Cite this article
Costanzo, M.R. Central Sleep Apnea in Patients with Heart Failure—How to Screen, How to Treat. Curr Heart Fail Rep 17, 277–287 (2020). https://doi.org/10.1007/s11897-020-00472-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-020-00472-0