
Abstract
Purpose of Review
Pulmonary hypertension due to left heart disease (PH-LHD) is the most common cause of pulmonary hypertension worldwide, yet therapies used to treat pulmonary arterial hypertension have failed to show efficacy in this population. Proper hemodynamic assessment and differentiation of pulmonary hypertension phenotypes is therefore critical for both current clinical practice and future research and therapeutic efforts.
Recent Findings
Substantial recent efforts have sought to improve the hemodynamic characterization of pulmonary hypertension for both diagnostic and prognostic purposes. These efforts include identifying occult LHD using provocative maneuvers as well as sub-classifying PH-LHD based on the presence or absence of a pre-capillary component. How to best define the pre-capillary component remains controversial as several studies have drawn conflicting conclusions. The lack of standardization of hemodynamic measurements as well as measurement fidelity concerns may explain some of the discrepant results. Non-hemodynamic methods of PH-LHD classification may also have an emerging role. Despite recent advances, therapeutic studies have largely remained disappointing.
Summary
In this review, we discuss the nuances and controversies surrounding diagnostic and prognostic hemodynamic characterization of PH-LHD as well as summarize the recent therapeutic efforts and ongoing challenges in this population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pulmonary hypertension (PH) associated with left heart disease (PH-LHD) is the most common form of PH worldwide and is associated with a worse prognosis than LHD without PH. Further, the presence of a pre-capillary component portends an even worse prognosis [1,2,3]. Despite its increasing recognition, there is lack of well-defined therapeutic options, and its hemodynamic assessment provides unique challenges. Here we review contemporary literature describing the recent advances in this field and the complexities in the hemodynamic assessment and management of PH-LHD.
Hemodynamic Definition and Classification of PH-LHD
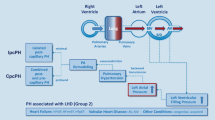
PH-LHD is defined as a mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg at rest, in the presence of elevated pulmonary arterial wedge pressure (PAWP) or left ventricular end diastolic pressure (LVEDP) > 15 mmHg [2, 4]. An increase in left-sided filling pressures differentiates this from World Health Organization (WHO) Group 1 pulmonary arterial hypertension (PAH). PH-LHD can be associated with heart failure with reduced ejection fraction (HFrEF), HF with preserved EF (HFpEF), left-sided valvular disease, or congenital cardiomyopathies [5].
An increase in PAWP initially causes a passive increase in pulmonary pressure or “isolated post-capillary PH (IpcPH)”, where the pulmonary pressures typically normalize with a reduction in PAWP. The diastolic pulmonary gradient (DPG = diastolic pulmonary artery pressure [dPAP] − PAWP), the transpulmonary gradient (TPG = mPAP − PAWP), and the pulmonary vascular resistance (PVR = TPG/cardiac output [CO]) are not significantly elevated (DPG < 7 mmHg, TPG < 12–15 mmHg and PVR < 3 Woods Unit [WU]) [2].
The prevailing paradigm is that persistent elevation in PAWP and ongoing heart failure provokes alveolar wall injury, pulmonary vasoconstriction, and sometimes, remodeling of the small resistance pulmonary arteries (PA) [6]. This leads to development of a pre-capillary component where pulmonary pressures increase out of proportion to PAWP. This has been coined “combined post- and pre-capillary PH (CpcPH)” and is characterized by an elevated DPG, TPG, and PVR. Interventions directed towards acute reduction in PAWP may not necessarily normalize pulmonary pressures [2], and CpcPH is associated with worse prognosis than IpcPH as well as LHD without PH [3]. Recent studies investigating hemodynamic classification and outcomes in PH-LHD are summarized in Table 1.
Diastolic Pulmonary Gradient and Updated Classification of PH-LHD
At the 5th World Symposium on PH in Nice, France, it was proposed that DPG should be the sole discriminator of IpcPH (DPG < 7 mmHg) and CpcPH (DPG ≥ 7 mmHg) [1]. This proposal was based on sound physiologic rationale: elevated left heart filling pressures lower pulmonary arterial compliance (PAC) independent of resistive load, increasing the pulmonary pulse pressure. As pulse pressure increases, mPAP increases out of proportion to dPAP, thereby indirectly raising the TPG and PVR [18]. The DPG is assessed during cardiac diastasis, eliminating contributions of flow state and the arterial Windkessel model. The dPAP is approximately equal to the PAWP and DPG is < 5 mmHg in most patients with LHD [8, 19].
Gerges et al. have demonstrated a worse prognosis in patients with PH-LHD and an elevated DPG ≥ 7 mmHg, both in the setting of an elevated TPG (≥ 12 mmHg) and when DPG is considered in isolation [7•, 20]. However, many other studies have failed to reproduce the prognostic value of DPG in both HFrEF and HFpEF [8, 9•, 10, 13, 21]. There are several factors that likely contribute to the discrepant results. First, as heart rate increases and diastole shortens, the gradient between dPAP and PAWP increases, irrespective of presence or absence of pre-capillary disease. Thus, significant tachycardia can raise the DPG [22]. Second, right ventricular (RV) function and adaptation to its afterload may have a more significant impact on prognosis than pulmonary vascular pressures (or disease) alone [23, 24]. Incorporation of flow (via stroke volume—a marker of ventricular function) may explain why PVR has proven to be a more robust prognostic marker than DPG or TPG. The fact that prognosis is also poorer in the setting of very low or negative DPG [9•] may suggest that LHD drives outcomes independent of pulmonary vascular disease. These data also remind us that the lack of a clear prognostic signal does not necessarily preclude the use of DPG as a diagnostic variable. Finally, the observation of frequent negative DPG values illustrates the challenges in accurately measuring the DPG [8, 9•, 12]. Lack of measurement standardization and artifact associated with use of fluid filled catheters undoubtedly contribute and will be discussed in sections below.
Recognizing some of the limitations of DPG, in 2015, European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines redefined CpcPH as mPAP ≥ 25 mmHg, mean PAWP > 15 mmHg, DPG ≥ 7 mmHg, and/or PVR > 3 Wood units. [4] There are ongoing concerns that the “or” portion of this definition can significantly raise the number of patients with CpcPH, based on an increase in PVR alone [25]. Additionally, Palazzini and colleagues did not find the new ERS/ERS definition predictive of worse outcome in their cohort [13•]. Most recently, Guazzi and Naeije have proposed defining CpcPH as DPG ≥ 7 mmHg and PVR > 3 Wood units, reasoning that an elevated DPG in the setting of a normal PVR is likely a false positive [23]. Whether this provides superior prognostication remains to be studied.
Despite the complexities in defining CpcPH, the data in totality show us that CpcPH has a worse prognosis as compared to Ipc-PH [13•] with a distinct pathophysiology. In an exploratory analysis of CpcPH patients, Assad et al. found genes and biological pathways in the lung known to contribute to PAH pathophysiology [11•]. Exercise breathing patterns in CpcPH are also more similar to PAH than IpcPH with a reduced prevalence of exercise oscillatory breathing [17]. Because of the outlined limitations of a purely hemodynamic definition, there is an ongoing need to develop biomarker or non-hemodynamic strategies to more precisely phenotype and accurately define CpcPH.
Pulmonary Arterial Compliance
PVR only describes the load imposed on the RV during steady-state blood flow. Pulmonary arterial compliance (PAC) is a second determinant of afterload that accounts for the pulsatile nature of blood flow. PAC specifically quantifies the distensibility of the pulmonary vasculature relative to changes in volume. PAC can be estimated a number of ways, most simply as stroke volume [SV]/PA pulse pressure. In the normal lung, PVR and PAC are inversely related and their product (RC time) is nearly constant. Thus, an increase in PVR is accompanied by a proportional decrease in PAC, over a wide range of severities of PH [26]. However, in LHD, an increase in PAWP further lowers PAC for any given PVR, thereby increasing RV pulsatile afterload [18]. Thus, in the unique setting of LHD, PAC bundles the effect of two hemodynamic measurements (PAWP and PVR), and is a more complete marker of total RV afterload. Not surprisingly, PAC is a superior prognostic marker to both PVR and DPG in predicting RV failure and adverse outcomes in PH-LHD [10•, 14, 27, 28]. It however is not helpful in differentiating IpcPH and CpcPH.
Hemodynamic Assessment of PH-LHD
Right heart catheterization (RHC) is the gold standard for diagnosis of PH. It is safe even in the setting of severe PH and is associated with low morbidity at experienced centers [4]. A RHC should always be performed before any PH-specific therapy is initiated. Preferred access sites include right internal jugular vein or antecubital vein, with the femoral vein as an alternative. Local anesthesia is used to facilitate line placement. Systemic sedation should generally be avoided, with preference for oral sedative medications, if needed. The patient remains supine with legs flat during the study. Blood pressure (BP) and systemic arterial oxygen saturation should be monitored. Care must be taken to properly flush pressure lines and ensure equipment calibration is up to date. The pressure transducer should be zeroed at the level of the left atrium which corresponds to the mid-thoracic line (halfway between the anterior aspect of the sternum and the table surface) [1, 29]. Pressures should always be accompanied by a simultaneous ECG recording and are recorded in the right atrium (RA), right ventricle (RV), pulmonary artery (PA), and the wedge positions while the patient is breathing spontaneously. Breath hold maneuvers are not recommended [30, 31]. Waveforms are analyzed at end-expiration when intra-thoracic pressures have the least effect on resting pressure measurements. A PAWP saturation (blood sample taken from the distal port while in the wedge position) should always be checked to ensure an adequate wedge, particularly when the PAWP is elevated. The PAWP saturation should be within 5% of the systemic oxygen saturation. An LVEDP should be measured when PAWP cannot be measured or the measured PAWP seems inconsistent with the clinical picture.
If tracings appear dampened (e.g., loss of a dicrotic notch in the PAP tracing or blunted RV end-diastolic pressure inflection point), catheter and tubing should be flushed to remove any air. Catheter whip may occur in high CO states and is minimized by relocating the catheter to a less turbulent area. More common than catheter whip, catheter ringing occurs as the heart rate approaches the inherent resonant frequency of the fluid filled catheter system. Microbubbles in the fluid-filled catheter exacerbate this issue by increasing the compliance of the system [32]. After thoroughly flushing the system, if catheter ringing is still present, a filter can be introduced [33] or more commonly a small amount of denser fluid (blood or contrast) can be added to the catheter [34]. However, the latter strategy can result in over-dampening of the waveform, with a resultant decrease in the sPAP and increase in the dPAP (Fig. 1a, b). This further illustrates the inherent limitation of using DPG as a diagnostic or prognostic marker in PH-LHD.

Pulmonary artery (PA) tracings from the same patient during a single right heart catheterization procedure. a A high degree of “ringing” artifact when using a fluid-filled catheter. Note the difficulty this poses in determining both the systolic and diastolic PA pressures. b PA tracing after a small amount of blood was aspirated into the catheter to change the compliance of the catheter. Note the reduced ringing artifact. However, now the systolic pressure is lower, and the diastolic pressure is higher. It is possible to over-dampen the tracing by this method, and a false elevation in the diastolic pressure may lead to a falsely elevated diastolic pulmonary gradient, misclassifying a patient as Cpc-PH. Note that the mean PA pressure is nearly the same in both tracings
Measurement of PAWP—Important Considerations and Caveats
The PAWP should be measured at end-diastole to reflect the LVEDP and confirmed with a wedge saturation, especially when the PAWP is elevated. Several publications have highlighted the lack of standardization of the PAWP measurement and the potential impact on pre-capillary parameters, especially the DPG [12, 15•, 35,36,37,38]. Use of the “mean”-PAWP value, which is averaged over the entire cardiac cycle and incorporates the V wave, results in lower or even negative DPG values when compared to measuring PAWP at end-diastole (typically as mean of the “a” wave or pre c-wave) and can significantly vary from measured LVEDP, leading to misclassification of PH [39]. This is particularly relevant in the presence of atrial fibrillation [35] and prominent V waves [12], where end-diastolic PAWP correlates best with LVEDP [40]. Wright and Mak recently proposed a novel approach to estimate PAWP near end-diastole with EKG gating [15•]. At least for the purposes of assessing for a pre-capillary component of PH where high accuracy and reproducibility in dPAP and PAWP is required, estimating PAWP at end-diastole appears to be the most appropriate technique. Perhaps just as relevant, they also described the impact of a fluid-filled catheter artifact on the dPAP [15•]. Whether more standardized approaches to measure the PAWP or the use of high-fidelity catheters to minimize artifact would improve the prognostic value of the DPG requires further study.
Estimation of Cardiac Output
The gold standard for estimating CO is the direct Fick method that involves measuring VO2 or oxygen consumption [CO = VO2 / systemic arteriovenous oxygen difference] [41]. As the required equipment for measuring oxygen consumption is cumbersome and not widely available, thermodilution (TD) [42] and the estimated Fick method are commonly used. TDCO should be measured in triplicate at end-expiration [4, 43]. Although the validity of TDCO has been questioned in the setting of low or high CO or severe tricuspid regurgitation [44,45,46], other studies have reported an agreement between TD and direct Fick, validating its use even under those circumstances [41, 47]. TDCO may be inaccurate in the presence of intra-cardiac shunts and should not be used in this circumstance. The estimated Fick method relies on estimating VO2 and is prone to error in heart failure, PH, or abnormal body habitus [48,49,50]. Discrepancies between different methods have a wide range of implications for the diagnosis and management of PH-LHD [51]. In an analysis of more than 15,000 adults undergoing RHC, TDCO and estimated Fick methods correlated poorly, varying by > 20% in a third of the patients. TDCO estimates were superior to estimated Fick in predicting all-cause mortality [52]. In the absence of an intra-cardiac shunt, TD is the recommended method to assess CO [30, 31].
Vasodilator Testing
Vasodilator testing is recommended during RHC in heart transplant candidates with CpcPH as absence of reversibility precludes eligibility for heart transplantation (HT) [53, 54]. The risk of early death presumably from RV failure is elevated when PVR > 5 WU or TPG > 15 mmHg [55, 56], and the risk increases incrementally with increasing PVR [57]. In a classic study of 293 patients undergoing evaluation for HT with vasodilator testing using intravenous (IV) nitroprusside [53], mortality was highest in patients whose PVR failed to drop below 2.5WU, followed by patients in whom the PVR was ≤ 2.5 WU, but nitroprusside administration resulted also in systemic hypotension (systolic BP (SBP) to < 85 mmHg). Short-term survival was similar in patients with an appropriate decrease in PVR without systemic hypotension compared with patients with normal PVR at baseline. Several subsequent studies have used various other vasodilators (nitroglycerin), inotropes (dobutamine, milrinone), inhaled nitric oxide (iNO), prostaglandin E1, inhaled or intravenous prostacyclin, and sildenafil for the evaluation of the reversibility of PH with varying effects on hemodynamics [58,59,60,61,62]. A contemporary meta-analysis revealed that sodium nitroprusside resulted in the most profound lowering of PAWP, while PAWP increased with iNO [63]. Nitroprusside and milrinone led to a most significant increase in cardiac output while prostacyclin and prostaglandin E1 exerted the most reduction in PVR.
The International Society for Heart and Lung Transplantation (ISHLT) recommends PVR > 5 WU or TPG > 15 mmHg as a relative contraindication to HT, although many centers consider extending this to a PVR > 3WU [54]. If PVR can be reduced to ≤ 2.5 WU without systemic hypotension, patients can be accepted as candidates. The guidelines do not provide recommendations on which vasodilator to use, and the choice is often determined by the center’s practice.
In general, for HT candidates with PVR > 3–5 WU and TPG > 15 mmHg, with elevated PAWP and systemic vascular resistance (SVR), and SBP > 90 mmHg, IV nitroprusside is our initial vasodilator of choice (prostaglandin E1 is an acceptable alternative). Elevation in PAWP alone can raise the PVR by multiple mechanisms [18, 64,65,66]. If PVR remains high despite lowering PAWP, we would consider addition of iNO or inhaled prostacyclin. In the absence of significantly elevated PAWP, and especially if CO is very low and mPAP is modestly elevated, we would consider the use of IV milrinone [58] with possible addition of iNO. In the absence of a reduced CO and normal or near normal PAWP, we consider the use of more selective pulmonary vasodilators including IV or inhaled prostacyclin, with preference for inhaled prostacyclin or iNO if SBP is low.
When an acute vasodilator challenge is unsuccessful, prolonged administration of vasodilators over 24 to 48 h maybe necessary. Some patients require longer duration of vasodilator therapy over days to weeks with serial RHC [67], and if still unsuccessful, are classified as irreversible PH-LHD, needing consideration for mechanical circulatory support (MCS) [2]. Most patients with “irreversible” PH-LHD still normalize PVR after MCS support, suggesting that a persistent “functional” component of PH exists in these patients [68,69,70] rather than significant pulmonary vascular remodeling.
In a retrospective analysis of the United Network of Organ Sharing (UNOS) data, an elevated DPG (using multiple cutoffs), even in combination with elevated PVR or TPG, failed to predict post-transplant mortality [8]. Using the updated classification of PH-LHD, Ghio et al. recently found that CpcPH patients had more significant improvement in PVR, TPG, and DPG during vasoreactive testing as compared to IpcPH patients [16]. Thus, the new classification of CpcPH does not help to identify irreversible PH-LHD. Al-Naamani and colleagues also found that vasoreactivity did not predict prognosis in their cohort of HFpEF patients with CpcPH [10•]. Currently, outside of assessing transplant candidacy, there is no clinical role for vasodilator testing in PH-LHD.
Provocative Testing
In the context of LHD, provocative testing during RHC with fluid challenge or exercise is helpful for evaluating exertional dyspnea of unknown origin with normal resting hemodynamics, in identifying early stages of LHD [71], and differentiating HFpEF from PAH in patients with normal PAWP at rest (Table 2) [77]. PAWP of course may be reduced to < 15 mmHg with diuresis or afterload reduction prior to RHC, leading to misclassification of PH [9•]. Provocative testing may also be helpful in identifying exercise induced PH (EIPH) [78, 79], preload insufficiency [80], and defining RV contractile reserve [81], though these latter topics are beyond the scope of the current review.
Fluid Challenge
It is important to remember that PAWP increases in healthy patients in response to a fluid bolus. However, patients with HFpEF exhibited a steeper increase in PAWP relative to the infused volume, as compared to healthy controls [82]. The volume and rate of fluid administration have clinical relevance, as slower infusions can lead to re-distribution into intra-vascular spaces [83], while more rapid infusions maybe poorly tolerated in HF [73, 82, 84]. In a retrospective analysis of 287 patients, including 202 patients labeled as non-group 2 PH, with mPAP ≥ 25 mmHg and PAWP ≤ 15 mmHg at rest, a fluid challenge of 500 cm3 of normal saline over 5–10 min led to a re-classification of 22% of patients as having occult pulmonary venous hypertension, based on an increase in PAWP to > 15 mmHg [73]. These patients had clinical, echocardiographic, and hemodynamic characteristics comparable to HFpEF patients, suggesting a distinct phenotype from PAH. However, a PAWP > 15 mmHg with fluid bolus can be seen in normal controls where a PAWP > 18 mmHg was not witnessed in healthy controls [63].
More recently, D’alto et al. studied 212 patients referred for RHC for evaluation of PH who underwent hemodynamic measurements before and after the administration of 7 ml/kg of saline over 5–10 min (~ 500 cm3) [72•]. After fluid administration, 8 and 6% of patients initially classified as no-PH and pre-capillary PH, respectively, were re-classified as post-capillary PH based on an increase in PAWP > 18 mmHg. Similar to prior studies [82], patients with PH-LHD had a steeper increase in PAWP, while the PAWP in patients without PH did not increase to > 18 mmHg. Interestingly, the DPG decreased with fluid challenge, as an acute rise in PAWP can be accompanied by a slower rise in dPAP due to the preserved distension of the resistive PAs [85].
The effect of pericardial constraint on ventricular interaction can also increase PAWP in response to a fluid challenge, in patients with normal hemodynamics [82] as well as RV pressure overload [83, 86]. RAP approximates pericardial pressure, and thus an increase in LV transmural pressure (PAWP − RAP) is suggestive of LHD as opposed to pericardial constraint. In the study by D’Alto et al., this increased only in patients with either overt or occult PH-LHD [72•].
In summary, an increase in PAWP > 18 mmHg in response to a fluid challenge with 500 cm3 of saline administered over 5–10 min may be useful to identify occult PH-LHD, particularly if accompanied increasing LV transmural pressure. Although exercise may be a more sensitive provocative test than fluid challenge to identify PH-LHD [84], the potential advantages of a fluid challenge include less variation with HR and blood pressure, less catheter artifact and dependence on patient’s exercise capacity, and the widespread availability of necessary equipment compared to what is required for exercise.
Exercise RHC
Exercise induces elevation in CO, PAWP, and/or PAP in normal subjects with increasing age [87], and those with HF or PH [71, 88, 89]. It is typical to perform an exercise RHC under a ramp protocol of incremental workloads (2–3 min per stage) with measurement of RAP, PAWP, mPAP, and CO at each stage [31]. Accurate leveling of the transducer is mandatory, and may be particularly challenging for upright exercise [31]. Bicycle exercise is preferred to upper extremity exercise, to avoid increased systemic vascular resistance with the latter. Fluid-filled catheters often result in excessive ringing and motion artifacts during exercise; therefore, only mean pressures should be measured. Large intra-thoracic pressure changes can occur during exercise leading to over-estimation of pressures if measured at end-expiration, especially in lung disease [90, 91]. Therefore, averaging the measurements over the respiratory cycle is recommended. Lastly, care must be taken to ensure an adequate PAWP. A recent position statement on exercise hemodynamics details many of these issues [31].
Exercise-Induced Changes in PAWP
An increase in PAWP of 10 mmHg or more may be observed with exercise in healthy controls [84, 89, 92, 93], with an average of 5 mmHg greater rise with supine vs upright exercise, although the absolute change in PAWP is similar [93]. However, there are several factors that affect this response. Exercise-induced increase in PAWP is more marked in older patients [76]. Changes in PAWP also vary with the duration and intensity of exercise. PAWP may exceed 20 mmHg early in exercise, but decline significantly within minutes [75•]. Such brief and early increases in PAWP may not be pathologic, and thus, measurements at multiple time points may be helpful. Exercise can also induce a ~ 2-fold greater increase in PAWP as compared to fluid challenge in patients with HFpEF, although exercise pressures were measured at end-expiration in this study [84]. In general, we consider an abnormal PAWP to be ≥ 25 mmHg with supine exercise or ≥ 20 mmHg with upright exercise, averaged over the entire respiratory cycle. In those over the age of 60, PAWP ≥ 25 has been reported in up to 30% of subjects apparently free of cardiovascular disease. Thus, exercise PAWP should be considered in the context of age [76], and age-specific definitions have also been proposed [94]. The effect of significant pericardial constraint should be excluded by considering the transmural LV pressure [83].
Exercise-Induced Changes in Cardiac Output
CO during exercise is best measured with the direct Fick method [31]. Oxygen saturations, hemoglobin concentration, and oxygen consumption should be measured. TDCO is considered an alternative if equipment for direct Fick is unavailable. However, the TD method can underestimate the CO as compared to the direct Fick method, particularly at higher outputs [74•].
Treatment Selection in PH-LHD
The primary treatment of PH-LHD is the management of the underlying LHD. There are no PAH-specific therapies currently approved for PH-LHD; rather, interventions are directed towards relieving symptoms of dyspnea and improving exercise capacity, or defining eligibility for HT [1, 2].
Treatment of PH-HFrEF
The cornerstone of treatment of PH-HFrEF is the use of guideline-directed therapies including beta-blockers (BB), angiotensin-converting enzyme inhibitors (ACE-I), or angiotensin receptor blockers (ARBs) or angiotensin receptor-neprilysin inhibitor (ARNI), aldosterone-antagonists, and hydralazine/nitrate combination [95, 96]. Relief of congestion with diuretics and vasodilators can improve PAPs and may require invasive monitoring [95, 97, 98]. Reducing left heart filling pressures alone may significantly lower PVR as discussed above. Non-pharmacologic therapies include consideration for cardiac resynchronization therapy when appropriate and MCS for suitable candidates [95]. Reversibility of fixed PH can occur with MCS, allowing candidacy for HT [99, 100].
PAH-specific therapies have been trialed in PH-LHD on the basis of PH being driven by increased endothelin-1 activity [101, 102] and impaired NO-dependent vasodilation [103]. However, studies involving parenteral prostacyclins [104] and endothelin receptor antagonists (ERA) [105,106,107,108] in HFrEF have demonstrated negative or neutral effects, and even trends towards harm [109, 110].
Sildenafil, a phosphodiesterase-5-inhibitor (PDE-5-I), promotes NO-dependent vasodilation by preventing the degradation of cyclic guanosine monophosphate (cGMP) [111]. In single-center studies, sildenafil has been shown to decrease TPG, increase CO [112], and improve exercise hemodynamics, VO2 [113], exercise capacity, and quality of life in HFrEF [114]. However, these studies used higher doses of sildenafil (25 to 75 mg three times daily [TID]) than what is approved for PAH therapy. A multicenter trial (SilHF, NCT01616381) to evaluate a lower dose of sildenafil in PH-HFrEF is currently ongoing. A retrospective study has suggested that sildenafil lowers mPAP and PVR in those with persistent PH after MCS implantation [115]. A randomized, placebo-controlled, multi-center study (SOPRANO, NCT02554903) is currently investigating the use of the ERA Macitentan in this clinical scenario.
Riociguat, a soluble guanylate cyclase (sGC) stimulator, sensitizes sGC to endogenous NO and directly stimulates sGC independent of NO, inducing vasodilation [116]. In the LEPHT trial, multiple doses of riociguat (0.5, 1, and 2 mg TID) were compared to placebo in 201 patients with PH-HFrEF. The study failed to meet the primary end point of reduction in mPAP after 16 weeks, but improved cardiac index and PVR [117]. Similarly, in SOCRATES-REDUCED, vericiguat did not meet the primary end point of reduction in NT-proBNP [118] compared with placebo in 456 patients with HFrEF (PH was not a requirement for study entry).
Treatment of PH-HFpEF
The management of HFpEF is limited to diuretics for the relief of volume overload and treatment of underlying conditions including hypertension, coronary artery disease, atrial fibrillation, and sleep apnea [95]. It is reasonable to control BPs with BB, ACE-I, or ARBs, although none of these drugs conclusively improve outcomes in HFpEF [95]. Aldosterone antagonists are recommended to reduce HF hospitalizations in eligible HFpEF patients [96, 119]. Nitrate therapy is ineffective in HFpEF to improve exercise tolerance or quality of life [120].
PAH-specific therapies have been studied in HFpEF with limited benefit. Sildenafil 50 mg TID improved hemodynamics and echocardiographic measures of RV function, in a placebo-controlled, single-centered trial of 44 patients with CpcPH in HFpEF at 6 months, with continued benefit at 1 year [121]. However, in a subsequent single-center study and in the RELAX study, sildenafil did not improve mean PAP, PAWP, CO, [122] exercise tolerance, or VO2 in HFpEF [122, 123]. Post hoc analysis from the RELAX study revealed that sildenafil failed to result in any significant reduction in RV afterload and likely reduced LV contractility [124]. The DILATE-1 trial compared multiple doses of riociguat (0.5, 1, and 2 mg) to placebo in 39 patients with PH-HFpEF including five with CpcPH. There was no significant change in mPAP at 6 h (primary end point) or PVR. Riociguat 2 mg as compared to placebo increased SV, lowered SVR, and decreased RV end-diastolic area, without increasing PAWP [125]. In another phase 2 study SOCRATES-PRESERVED, vericiguat did not change the primary end points of NT-proBNP and left atrial volume at 12 weeks compared with placebo in 477 patients with HFpEF (presence of PH was not an inclusion criteria), although there were some improvements in quality of life metrics [126].
Recent trials of ERAs in HFpEF have been largely disappointing. A selective endothelin A receptor antagonist, sitaxsentan, improved treadmill exercise time compared with placebo in 192 patients with HFpEF after 6 months of therapy, without an improvement in LV mass or diastolic indices [127]. The BADDHY Trial evaluated 12 weeks of therapy with bosentan vs placebo in 20 patients with PH-HFpEF, including 4 with CpcPH, demonstrated worsening PAP and RAP with bosentan [128]. The MELODY Trial evaluating macitentan in 63 patients with CpcPH, LVEF > 30%, most of whom had HFpEF, demonstrated no significant difference in the primary end point of worsening functional class or fluid retention, although more patients receiving macitentan had worsening fluid retention, without significant reduction in RAP or PVR after 12 weeks of treatment [129] compared to placebo. This study is particularly notable since its careful inclusion criteria led to selection of subjects with clear pre-capillary disease (average PVR of 5.8 WU, TPG of 27 mmHg, and DPG of 10 mmHg.), regardless of the definition used.
The recent clinical trials in both HFrEF-PH and HFpEF-PH remind us that the use of PAH-specific therapies in PH-LHD should only be in the context of clinical trials.
Conclusions and Future Directions
In recent years, there has been significant progress in the classification and characterization of PH-LHD including identification of a subset who merit special attention (CpcPH). The significance of accurate hemodynamic assessment for its diagnosis and prognosis cannot be overstated. Currently, there are currently no approved PH-specific therapies in the setting of LHD, and treatment efforts remain limited to targeting underlying left heart disease. Future research is needed to improve both hemodynamic and non-hemodynamic characterization of PH-LHD phenotypes as well as novel therapeutic strategies to target these populations.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Vachiery JL, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100–8. https://doi.org/10.1016/j.jacc.2013.10.033.
Fang JC, DeMarco T, Givertz MM, Borlaug BA, Lewis GD, Rame JE, et al. World Health Organization Pulmonary Hypertension Group 2: pulmonary hypertension due to left heart disease in the adult—a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2012;31(9):913–33. https://doi.org/10.1016/j.healun.2012.06.002.
Guazzi M, Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation. 2012;126(8):975–90. https://doi.org/10.1161/CIRCULATIONAHA.111.085761.
Galie N, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. https://doi.org/10.1093/eurheartj/ehv317.
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–41. https://doi.org/10.1016/j.jacc.2013.10.029.
Delgado JF, Conde E, Sánchez V, López-Ríos F, Gómez-Sánchez MA, Escribano P, et al. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail. 2005;7(6):1011–6. https://doi.org/10.1016/j.ejheart.2004.10.021.
• Gerges C, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in "out-of-proportion" pulmonary hypertension. Chest. 2013;143(3):758–66. This study found that in patients with postcapillary PH and TPG >12mmHg, DPG > 7mmHg was associated with a worse median survival. In 18 patients, lung tissue in patients with elevated DPG was evaluated and showed advanced remodeling of the vasculature.
Tedford RJ, Beaty CA, Mathai SC, Kolb TM, Damico R, Hassoun PM, et al. Prognostic value of the pre-transplant diastolic pulmonary artery pressure-to-pulmonary capillary wedge pressure gradient in cardiac transplant recipients with pulmonary hypertension. J Heart Lung Transplant. 2014;33(3):289–97. https://doi.org/10.1016/j.healun.2013.11.008.
• Tampakakis E, et al. The diastolic pulmonary gradient does not predict survival in patients with pulmonary hypertension due to left heart disease. JACC Heart Fail. 2015;3(1):9–16. In this retrospective, largely HFrEF cohort, DPG was not associated with mortality in PH-LHD. TPG and PVR both predicted mortality. 36% of PH-LHD patients had a negative DPG.
• Al-Naamani N, et al. Pulmonary arterial capacitance is an important predictor of mortality in heart failure with a preserved ejection fraction. JACC Heart Fail. 2015;3(6):467–74. This study found that pulmonary vascular compliance was the best predictor of mortality in a PH-HFpEF cohort. DPG did not predict outcome, and acute vasodilator response was not associated with improved survival (inhaled NO).
• Assad TR, et al. Clinical and biological insights into combined post- and pre-capillary pulmonary hypertension. J Am Coll Cardiol. 2016;68(23):2525–36. Patients with CpcPH had genetic polymorphisms shared with PAH that were not present in Ipc-PH patients in this novel study.
Nagy AI, Venkateshvaran A, Merkely B, Lund LH, Manouras A. Determinants and prognostic implications of the negative diastolic pulmonary pressure gradient in patients with pulmonary hypertension due to left heart disease. Eur J Heart Fail. 2017;19(1):88–97. https://doi.org/10.1002/ejhf.675.
• Palazzini, M., et al., Pulmonary hypertension due to left heart disease: analysis of survival according to the haemodynamic classification of the 2015 ESC/ERS guidelines and insights for future changes. Eur J Heart Fail, 2017. In PH-LHD patients, using the revised ESC/ERS guidelines, no difference in survival was noted between CpcPH and Ipc-PH. PVR had a better predictive value than DPG in PH-LHD patients.
Adir Y, Guazzi M, Offer A, Temporelli PL, Cannito A, Ghio S. Pulmonary hemodynamics in heart failure patients with reduced or preserved ejection fraction and pulmonary hypertension: similarities and disparities. Am Heart J. 2017;192:120–7. https://doi.org/10.1016/j.ahj.2017.06.006.
• Wright SP, et al. Diastolic pressure difference to classify pulmonary hypertension in the assessment of heart transplant candidates. Circ Heart Fail, 2017. 10(9). This study employed QRS-gating methods to assess the PAWP, which led to overall higher DPG values, a greater proportion of CpcPH patients, and fewer negative DPG values compared to usual methods.
Ghio, S., Crimi G., Temporelli P.L., Traversi E., la Rovere M.T., Cannito A., Vizza D., Scelsi L., Raineri C., Guazzi M., Oltrona Visconti L., Haemodynamic effects of an acute vasodilator challenge in heart failure patients with reduced ejection fraction and different forms of post-capillary pulmonary hypertension. Eur J Heart Fail, 2017. https://doi.org/10.1002/ejhf.1067.
Caravita S, Faini A, Deboeck G, Bondue A, Naeije R, Parati G, et al. Pulmonary hypertension and ventilation during exercise: role of the pre-capillary component. J Heart Lung Transplant. 2017;36(7):754–62. https://doi.org/10.1016/j.healun.2016.12.011.
Tedford RJ, Hassoun PM, Mathai SC, Girgis RE, Russell SD, Thiemann DR, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation. 2012;125(2):289–97. https://doi.org/10.1161/CIRCULATIONAHA.111.051540.
Naeije R, Vachiery JL, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41(1):217–23. https://doi.org/10.1183/09031936.00074312.
Gerges M, Gerges C, Pistritto AM, Lang MB, Trip P, Jakowitsch J, et al. Pulmonary hypertension in heart failure. Epidemiology, right ventricular function, and survival. Am J Respir Crit Care Med. 2015;192(10):1234–46. https://doi.org/10.1164/rccm.201503-0529OC.
Mazimba S, Kennedy JLW, Zhuo D, Bergin J, Abuannadi M, Tallaj J, et al. Decreased pulmonary arterial proportional pulse pressure after pulmonary artery catheter optimization for advanced heart failure is associated with adverse clinical outcomes. J Card Fail. 2016;22(12):954–61. https://doi.org/10.1016/j.cardfail.2016.03.019.
Enson Y, Wood JA, Mantaras NB, Harvey RM. The influence of heart rate on pulmonary arterial-left ventricular pressure relationships at end-diastole. Circulation. 1977;56(4 Pt 1):533–9. https://doi.org/10.1161/01.CIR.56.4.533.
Guazzi M, Naeije R. Pulmonary hypertension in heart failure: Pathophysiology, Pathobiology, and Emerging Clinical Perspectives. J Am Coll Cardiol. 2017;69(13):1718–34. https://doi.org/10.1016/j.jacc.2017.01.051.
Naeije R. Measurement to predict survival: the case of diastolic pulmonary gradient. JACC Heart Fail. 2015;3(5):425. https://doi.org/10.1016/j.jchf.2014.12.014.
Gerges M, Gerges C, Lang IM. How to define pulmonary hypertension due to left heart disease. Eur Respir J. 2016;48(2):553–5. https://doi.org/10.1183/13993003.00432-2016.
Lankhaar JW, Westerhof N, Faes TJC, Tji-Joong Gan C, Marques KM, Boonstra A, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J. 2008;29(13):1688–95. https://doi.org/10.1093/eurheartj/ehn103.
Dupont M, Mullens W, Skouri HN, Abrahams Z, Wu Y, Taylor DO, et al. Prognostic role of pulmonary arterial capacitance in advanced heart failure. Circ Heart Fail. 2012;5(6):778–85. https://doi.org/10.1161/CIRCHEARTFAILURE.112.968511.
Pellegrini P, Rossi A, Pasotti M, Raineri C, Cicoira M, Bonapace S, et al. Prognostic relevance of pulmonary arterial compliance in patients with chronic heart failure. Chest. 2014;145(5):1064–70. https://doi.org/10.1378/chest.13-1510.
Kovacs G, Avian A, Olschewski A, Olschewski H. Zero reference level for right heart catheterisation. Eur Respir J. 2013;42(6):1586–94. https://doi.org/10.1183/09031936.00050713.
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–50. https://doi.org/10.1016/j.jacc.2013.10.032.
Kovacs G, Herve P, Barbera JA, Chaouat A, Chemla D, Condliffe R, et al. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J. 2017;50(5):1700578. https://doi.org/10.1183/13993003.00578-2017.
Scruggs V, Pietras RJ, Rosen KM. Frequency response of fluid-filled catheter-micromanometer systems used for measurement of left ventricular pressure. Am Heart J. 1975;89(5):619–24. https://doi.org/10.1016/0002-8703(75)90508-6.
Brower RW, Spaans W, Rewiersma PA, Meester GT. A fully automatic device for compensating for artifacts in conventional catheter-manometer pressure recordings. Biomed Eng. 1975;10(8):305–10.
Lim, M., et al., Hemodynamic rounds: interpretation of cardiac pathophysiology from pressure waveform analysis, 3rd edition, ed. M. Kern. 2009: John Wiley & Sons, Inc.
Dickinson MG, Lam CS, Rienstra M, Vonck TE, Hummel YM, Voors AA, et al. Atrial fibrillation modifies the association between pulmonary artery wedge pressure and left ventricular end-diastolic pressure. Eur J Heart Fail. 2017;19(11):1483–90. https://doi.org/10.1002/ejhf.959.
Tampakakis E, Tedford RJ. Balancing the positives and negatives of the diastolic pulmonary gradient. Eur J Heart Fail. 2017;19(1):98–100. https://doi.org/10.1002/ejhf.704.
Houston BA, Tedford RJ. Is pulmonary artery wedge pressure a Fib in A-Fib? Eur J Heart Fail. 2017;19(11):1491–4. https://doi.org/10.1002/ejhf.992.
Houston BA, Tedford RJ. What we talk about when we talk about the wedge pressure. Circ Heart Fail. 2017;10(9):e004450. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004450.
Halpern SD, Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest. 2009;136(1):37–43. https://doi.org/10.1378/chest.08-2784.
Braunwald E, Frahm CJ, Ross J Jr. Studies on Starling's law of the heart. V. Left ventricular function in man. J Clin Invest. 1961;40(10):1882–90. https://doi.org/10.1172/JCI104412.
Hoeper MM, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med. 1999;160(2):535–41. https://doi.org/10.1164/ajrccm.160.2.9811062.
Ganz W, Donoso R, Marcus HS, Forrester JS, Swan HJC. A new technique for measurement of cardiac output by thermodilution in man. Am J Cardiol. 1971;27(4):392–6. https://doi.org/10.1016/0002-9149(71)90436-X.
Stevens JH, Raffin TA, Mihm FG, Rosenthal MH, Stetz CW. Thermodilution cardiac output measurement. Effects of the respiratory cycle on its reproducibility. JAMA. 1985;253(15):2240–2. https://doi.org/10.1001/jama.1985.03350390082030.
Nishikawa T, Dohi S. Errors in the measurement of cardiac output by thermodilution. Can J Anaesth. 1993;40(2):142–53. https://doi.org/10.1007/BF03011312.
Hillis LD, Firth BG, Winniford MD. Analysis of factors affecting the variability of Fick versus indicator dilution measurements of cardiac output. Am J Cardiol. 1985;56(12):764–8. https://doi.org/10.1016/0002-9149(85)91132-4.
Cigarroa RG, Lange RA, Williams RH, Bedotto JB, Hillis LD. Underestimation of cardiac output by thermodilution in patients with tricuspid regurgitation. Am J Med. 1989;86(4):417–20. https://doi.org/10.1016/0002-9343(89)90339-2.
Yung GL, Fedullo PF, Kinninger K, Johnson W, Channick RN. Comparison of impedance cardiography to direct Fick and thermodilution cardiac output determination in pulmonary arterial hypertension. Congest Heart Fail. 2004;10(2 Suppl 2):7–10. https://doi.org/10.1111/j.1527-5299.2004.03406.x.
Fakler U, Pauli C, Hennig M, Sebening W, Hess J. Assumed oxygen consumption frequently results in large errors in the determination of cardiac output. J Thorac Cardiovasc Surg. 2005;130(2):272–6. https://doi.org/10.1016/j.jtcvs.2005.02.048.
Narang N, Gore MO, Snell PG, Ayers CR, Lorenzo S, Carrick-Ranson G, et al. Accuracy of estimating resting oxygen uptake and implications for hemodynamic assessment. Am J Cardiol. 2012;109(4):594–8. https://doi.org/10.1016/j.amjcard.2011.10.010.
Narang N, Thibodeau JT, Levine BD, Gore MO, Ayers CR, Lange RA, et al. Inaccuracy of estimated resting oxygen uptake in the clinical setting. Circulation. 2014;129(2):203–10. https://doi.org/10.1161/CIRCULATIONAHA.113.003334.
Fares WH, Blanchard SK, Stouffer GA, Chang PP, Rosamond WD, Ford HJ, et al. Thermodilution and Fick cardiac outputs differ: impact on pulmonary hypertension evaluation. Can Respir J. 2012;19(4):261–6. https://doi.org/10.1155/2012/261793.
Opotowsky AR, Hess E, Maron BA, Brittain EL, Barón AE, Maddox TM, et al. Thermodilution vs estimated Fick cardiac output measurement in clinical practice: an analysis of mortality from the Veterans Affairs Clinical Assessment, Reporting, and Tracking (VA CART) program and Vanderbilt University. JAMA Cardiol. 2017;2(10):1090–9. https://doi.org/10.1001/jamacardio.2017.2945.
Costard-Jackle A, Fowler MB. Influence of preoperative pulmonary artery pressure on mortality after heart transplantation: testing of potential reversibility of pulmonary hypertension with nitroprusside is useful in defining a high risk group. J Am Coll Cardiol. 1992;19(1):48–54. https://doi.org/10.1016/0735-1097(92)90050-W.
Mehra MR, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates—2006. J Heart Lung Transplant. 2006;25(9):1024–42. https://doi.org/10.1016/j.healun.2006.06.008.
Griepp RB, Stinson EB, Dong E, Clark DA, Shumway NE. Determinants of operative risk in human heart transplantation. Am J Surg. 1971;122(2):192–7. https://doi.org/10.1016/0002-9610(71)90316-3.
Murali S, Kormos RL, Uretsky BF, Schechter D, Reddy PS, Denys BG, et al. Preoperative pulmonary hemodynamics and early mortality after orthotopic cardiac transplantation: the Pittsburgh experience. Am Heart J. 1993;126(4):896–904. https://doi.org/10.1016/0002-8703(93)90704-D.
Kirklin JK, Naftel DC, Kirklin JW, Blackstone EH, White-Williams C, Bourge RC. Pulmonary vascular resistance and the risk of heart transplantation. J Heart Transplant. 1988;7(5):331–6.
Givertz MM, Hare JM, Loh E, Gauthier DF, Colucci WS. Effect of bolus milrinone on hemodynamic variables and pulmonary vascular resistance in patients with severe left ventricular dysfunction: a rapid test for reversibility of pulmonary hypertension. J Am Coll Cardiol. 1996;28(7):1775–80. https://doi.org/10.1016/S0735-1097(96)00399-3.
von Scheidt W, et al. Prostaglandin E1 testing in heart failure-associated pulmonary hypertension enables transplantation: the PROPHET study. J Heart Lung Transplant. 2006;25(9):1070–6.
Loh E, Stamler JS, Hare JM, Loscalzo J, Colucci WS. Cardiovascular effects of inhaled nitric oxide in patients with left ventricular dysfunction. Circulation. 1994;90(6):2780–5. https://doi.org/10.1161/01.CIR.90.6.2780.
Alaeddini J, Uber PA, Park MH, Scott RL, Ventura HO, Mehra MR. Efficacy and safety of sildenafil in the evaluation of pulmonary hypertension in severe heart failure. Am J Cardiol. 2004;94(11):1475–7. https://doi.org/10.1016/j.amjcard.2004.07.157.
Weston MW, Isaac BF, Crain C. The use of inhaled prostacyclin in nitroprusside-resistant pulmonary artery hypertension. J Heart Lung Transplant. 2001;20(12):1340–4. https://doi.org/10.1016/S1053-2498(01)00320-5.
Guglin M, Mehra S, Mason TJ. Comparison of drugs for pulmonary hypertension reversibility testing: a meta-analysis. Pulm Circ. 2013;3(2):406–13. https://doi.org/10.4103/2045-8932.113180.
West JB, Dollery CT, Heard BE. Increased pulmonary vascular resistance in the dependent zone of the isolated dog lung caused by perivascular edema. Circ Res. 1965;17(3):191–206. https://doi.org/10.1161/01.RES.17.3.191.
Melenovsky V, Andersen MJ, Andress K, Reddy YN, Borlaug BA. Lung congestion in chronic heart failure: haemodynamic, clinical, and prognostic implications. Eur J Heart Fail. 2015;17(11):1161–71. https://doi.org/10.1002/ejhf.417.
Barnett CF, De Marco T. Pulmonary hypertension associated with left-sided heart disease. Heart Fail Clin. 2012;8(3):447–59. https://doi.org/10.1016/j.hfc.2012.04.009.
Al-Kindi SG, Farhoud M, Zacharias M, Ginwalla MB, ElAmm CA, Benatti RD, et al. Left ventricular assist devices or inotropes for decreasing pulmonary vascular resistance in patients with pulmonary hypertension listed for heart transplantation. J Card Fail. 2017;23(3):209–15. https://doi.org/10.1016/j.cardfail.2016.06.421.
Mikus E, Stepanenko A, Krabatsch T, Loforte A, Dandel M, Lehmkuhl HB, et al. Reversibility of fixed pulmonary hypertension in left ventricular assist device support recipients. Eur J Cardiothorac Surg. 2011;40(4):971–7. https://doi.org/10.1016/j.ejcts.2011.01.019.
Alba AC, Rao V, Ross HJ, Jensen AS, Sander K, Gustafsson F, et al. Impact of fixed pulmonary hypertension on postheart transplant outcomes in bridge-to-transplant patients. J Heart Lung Transplant. 2010;29(11):1253–8. https://doi.org/10.1016/j.healun.2010.06.002.
Masri SC, Tedford RJ, Colvin MM, Leary PJ, Cogswell R. Pulmonary arterial compliance improves rapidly after left ventricular assist device implantation. ASAIO J. 2017;63(2):139–43. https://doi.org/10.1097/MAT.0000000000000467.
Borlaug BA, Nishimura RA, Sorajja P, Lam CSP, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3(5):588–95. https://doi.org/10.1161/CIRCHEARTFAILURE.109.930701.
• D'Alto M, et al. Clinical relevance of fluid challenge in patients evaluated for pulmonary hypertension. Chest. 2017;151(1):119–26. Using a cutoff of 18mmHg for the PAWP, fluid challenge with 7 mL/kg of saline allowed reclassication of 6–8% of patients from precapillary PH or normal to PH-LHD.
Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail. 2014;7(1):116–22. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000468.
• Hsu S, et al. Use of thermodilution cardiac output overestimates diagnoses of exercise-induced pulmonary hypertension. Pulm Circ. 2017;7(1):253–5. In this study, thermodilution underestimated cardiac output during exercise compared with direct Fick.
• Wright SP, et al. The pulmonary artery wedge pressure response to sustained exercise is time-variant in healthy adults. Heart. 2016;102(6):438–43. This study described the pulmonary pressure response to sustained, submaximal exercise in healthy volunteers, finding that PAWP may routinely increase to >20mmHg early in exercise, and that PAWP and PA pressures may decline during continued exercise.
Wolsk E, Bakkestrøm R, Thomsen JH, Balling L, Andersen MJ, Dahl JS, et al. The influence of age on hemodynamic parameters during rest and exercise in healthy individuals. JACC Heart Fail. 2017;5(5):337–46. https://doi.org/10.1016/j.jchf.2016.10.012.
Thenappan T, Shah SJ, Gomberg-Maitland M, Collander B, Vallakati A, Shroff P, et al. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail. 2011;4(3):257–65. https://doi.org/10.1161/CIRCHEARTFAILURE.110.958801.
Tolle JJ, Waxman AB, van Horn TL, Pappagianopoulos PP, Systrom DM. Exercise-induced pulmonary arterial hypertension. Circulation. 2008;118(21):2183–9. https://doi.org/10.1161/CIRCULATIONAHA.108.787101.
Mullin CJ, Hsu S, Amancherla K, Wand A, Rhodes P, Leary PJ, et al. Evaluation of criteria for exercise-induced pulmonary hypertension in patients with resting pulmonary hypertension. Eur Respir J. 2017;50(3):1700784. https://doi.org/10.1183/13993003.00784-2017.
Oldham WM, Lewis GD, Opotowsky AR, Waxman AB, Systrom DM. Unexplained exertional dyspnea caused by low ventricular filling pressures: results from clinical invasive cardiopulmonary exercise testing. Pulm Circ. 2016;6(1):55–62. https://doi.org/10.1086/685054.
Hsu S, Houston BA, Tampakakis E, Bacher AC, Rhodes PS, Mathai SC, et al. Right ventricular functional reserve in pulmonary arterial hypertension. Circulation. 2016;133(24):2413–22. https://doi.org/10.1161/CIRCULATIONAHA.116.022082.
Fujimoto N, Borlaug BA, Lewis GD, Hastings JL, Shafer KM, Bhella PS, et al. Hemodynamic responses to rapid saline loading: the impact of age, sex, and heart failure. Circulation. 2013;127(1):55–62. https://doi.org/10.1161/CIRCULATIONAHA.112.111302.
Borlaug BA. Invasive assessment of pulmonary hypertension: time for a more fluid approach? Circ Heart Fail. 2014;7(1):2–4. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000983.
Andersen MJ, Olson TP, Melenovsky V, Kane GC, Borlaug BA. Differential hemodynamic effects of exercise and volume expansion in people with and without heart failure. Circ Heart Fail. 2015;8(1):41–8. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001731.
Naeije R, D'Alto M. The diagnostic challenge of group 2 pulmonary hypertension. Prog Cardiovasc Dis. 2016;59(1):22–9. https://doi.org/10.1016/j.pcad.2016.05.003.
Tyberg JV, Taichman GC, Smith ER, Douglas NW, Smiseth OA, Keon WJ. The relationship between pericardial pressure and right atrial pressure: an intraoperative study. Circulation. 1986;73(3):428–32. https://doi.org/10.1161/01.CIR.73.3.428.
Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888–94. https://doi.org/10.1183/09031936.00145608.
Maeder MT, Thompson BR, Brunner-la Rocca HP, Kaye DM. Hemodynamic basis of exercise limitation in patients with heart failure and normal ejection fraction. J Am Coll Cardiol. 2010;56(11):855–63. https://doi.org/10.1016/j.jacc.2010.04.040.
Lewis GD, Murphy RM, Shah RV, Pappagianopoulos PP, Malhotra R, Bloch KD, et al. Pulmonary vascular response patterns during exercise in left ventricular systolic dysfunction predict exercise capacity and outcomes. Circ Heart Fail. 2011;4(3):276–85. https://doi.org/10.1161/CIRCHEARTFAILURE.110.959437.
Boerrigter BG, Waxman AB, Westerhof N, Vonk-Noordegraaf A, Systrom DM. Measuring central pulmonary pressures during exercise in COPD: how to cope with respiratory effects. Eur Respir J. 2014;43(5):1316–25. https://doi.org/10.1183/09031936.00016913.
Lewis GD, Bossone E, Naeije R, Grunig E, Saggar R, Lancellotti P, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation. 2013;128(13):1470–9. https://doi.org/10.1161/CIRCULATIONAHA.112.000667.
Bevegard S, Holmgren A, Jonsson B. Circulatory studies in well trained athletes at rest and during heavy exercise. With special reference to stroke volume and the influence of body position. Acta Physiol Scand. 1963;57(1-2):26–50. https://doi.org/10.1111/j.1748-1716.1963.tb02572.x.
Thadani U, Parker JO. Hemodynamics at rest and during supine and sitting bicycle exercise in normal subjects. Am J Cardiol. 1978;41(1):52–9. https://doi.org/10.1016/0002-9149(78)90131-5.
Oliveira RK, et al. Age-related upper limits of normal for maximum upright exercise pulmonary haemodynamics. Eur Respir J. 2016;47(4):1179–88. https://doi.org/10.1183/13993003.01307-2015.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):1810–52. https://doi.org/10.1161/CIR.0b013e31829e8807.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey d Jr, Colvin MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136(6):e137–61. https://doi.org/10.1161/CIR.0000000000000509.
Binanay C, Califf RM, Hasselblad V, O'Connor CM, Shah MR, Sopko G, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294(13):1625–33. https://doi.org/10.1001/jama.294.13.1625.
Abraham WT, Adamson PB, Bourge RC, Aaron MF, Costanzo MR, Stevenson LW, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet. 2011;377(9766):658–66. https://doi.org/10.1016/S0140-6736(11)60101-3.
Haddad H, Elabbassi W, Moustafa S, Davies R, Mesana T, Hendry P, et al. Left ventricular assist devices as bridge to heart transplantation in congestive heart failure with pulmonary hypertension. ASAIO J. 2005;51(4):456–60. https://doi.org/10.1097/01.mat.0000169125.21268.d7.
Zimpfer D, Zrunek P, Roethy W, Czerny M, Schima H, Huber L, et al. Left ventricular assist devices decrease fixed pulmonary hypertension in cardiac transplant candidates. J Thorac Cardiovasc Surg. 2007;133(3):689–95. https://doi.org/10.1016/j.jtcvs.2006.08.104.
Givertz MM, Colucci WS, LeJemtel TH, Gottlieb SS, Hare JM, Slawsky MT, et al. Acute endothelin A receptor blockade causes selective pulmonary vasodilation in patients with chronic heart failure. Circulation. 2000;101(25):2922–7. https://doi.org/10.1161/01.CIR.101.25.2922.
Spieker LE, Mitrovic V, Noll G, Pacher R, Schulze MR, Muntwyler J, et al. Acute hemodynamic and neurohumoral effects of selective ET(A) receptor blockade in patients with congestive heart failure. ET 003 investigators. J Am Coll Cardiol. 2000;35(7):1745–52. https://doi.org/10.1016/S0735-1097(00)00649-5.
Porter TR, Taylor DO, Cycan A, Fields J, Bagley CW, Pandian NG, et al. Endothelium-dependent pulmonary artery responses in chronic heart failure: influence of pulmonary hypertension. J Am Coll Cardiol. 1993;22(5):1418–24. https://doi.org/10.1016/0735-1097(93)90552-C.
Califf RM, Adams KF, McKenna WJ, Gheorghiade M, Uretsky BF, McNulty SE, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44–54. https://doi.org/10.1016/S0002-8703(97)70105-4.
Packer M, McMurray J, Massie BM, Caspi A, Charlon V, Cohen-Solal A, et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J Card Fail. 2005;11(1):12–20. https://doi.org/10.1016/j.cardfail.2004.05.006.
Luscher TF, et al. Hemodynamic and neurohumoral effects of selective endothelin A (ET(A)) receptor blockade in chronic heart failure: the Heart Failure ET(A) Receptor Blockade Trial (HEAT). Circulation. 2002;106(21):2666–72. https://doi.org/10.1161/01.CIR.0000038497.80095.E1.
Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364(9431):347–54. https://doi.org/10.1016/S0140-6736(04)16723-8.
McMurray JJ, et al. Effects of tezosentan on symptoms and clinical outcomes in patients with acute heart failure: the VERITAS randomized controlled trials. JAMA. 2007;298(17):2009–19. https://doi.org/10.1001/jama.298.17.2009.
Humbert M, et al. Pulmonary edema complicating continuous intravenous prostacyclin in pulmonary capillary hemangiomatosis. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1681–5. https://doi.org/10.1164/ajrccm.157.5.9708065.
Preston IR, Klinger JR, Houtchens J, Nelson D, Mehta S, Hill NS. Pulmonary edema caused by inhaled nitric oxide therapy in two patients with pulmonary hypertension associated with the CREST syndrome. Chest. 2002;121(2):656–9. https://doi.org/10.1378/chest.121.2.656.
Katz SD, Balidemaj K, Homma S, Wu H, Wang J, Maybaum S. Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow-mediated vasodilation in patients with chronic heart failure. J Am Coll Cardiol. 2000;36(3):845–51. https://doi.org/10.1016/S0735-1097(00)00790-7.
Jabbour A, Keogh A, Hayward C, Macdonald P. Chronic sildenafil lowers transpulmonary gradient and improves cardiac output allowing successful heart transplantation. Eur J Heart Fail. 2007;9(6–7):674–7. https://doi.org/10.1016/j.ejheart.2007.01.008.
Lewis GD, Lachmann J, Camuso J, Lepore JJ, Shin J, Martinovic ME, et al. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation. 2007;115(1):59–66. https://doi.org/10.1161/CIRCULATIONAHA.106.626226.
Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116(14):1555–62. https://doi.org/10.1161/CIRCULATIONAHA.107.716373.
Tedford RJ, Hemnes AR, Russell SD, Wittstein IS, Mahmud M, Zaiman AL, et al. PDE5A inhibitor treatment of persistent pulmonary hypertension after mechanical circulatory support. Circulation Heart failure. 2008;1(4):213–9. https://doi.org/10.1161/CIRCHEARTFAILURE.108.796789.
Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, Bolkow D, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33(4):785–92. https://doi.org/10.1183/09031936.00039808.
Bonderman D, Ghio S, Felix SB, Ghofrani HA, Michelakis E, Mitrovic V, et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. 2013;128(5):502–11. https://doi.org/10.1161/CIRCULATIONAHA.113.001458.
Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CSP, Maggioni AP, et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES-REDUCED randomized trial. JAMA. 2015;314(21):2251–62. https://doi.org/10.1001/jama.2015.15734.
Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383–92. https://doi.org/10.1056/NEJMoa1313731.
Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH, et al. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med. 2015;373(24):2314–24. https://doi.org/10.1056/NEJMoa1510774.
Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124(2):164–74. https://doi.org/10.1161/CIRCULATIONAHA.110.983866.
Hoendermis ES, Liu LCY, Hummel YM, van der Meer P, de Boer RA, Berger RMF, et al. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36(38):2565–73. https://doi.org/10.1093/eurheartj/ehv336.
Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268–77. https://doi.org/10.1001/jama.2013.2024.
Borlaug BA, Lewis GD, McNulty SE, Semigran MJ, LeWinter M, Chen H, et al. Effects of sildenafil on ventricular and vascular function in heart failure with preserved ejection fraction. Circ Heart Fail. 2015;8(3):533–41. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001915.
Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014;146(5):1274–85. https://doi.org/10.1378/chest.14-0106.
Filippatos G, Maggioni AP, Lam CSP, Pieske-Kraigher E, Butler J, Spertus J, et al. Patient-reported outcomes in the SOluble guanylate cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. Eur J Heart Fail. 2017;19(6):782–91. https://doi.org/10.1002/ejhf.800.
Zile MR, Bourge RC, Redfield MM, Zhou D, Baicu CF, Little WC. Randomized, double-blind, placebo-controlled study of sitaxsentan to improve impaired exercise tolerance in patients with heart failure and a preserved ejection fraction. JACC Heart Fail. 2014;2(2):123–30. https://doi.org/10.1016/j.jchf.2013.12.002.
Koller B, Steringer-Mascherbauer R, Ebner CH, Weber T, Ammer M, Eichinger J, et al. Pilot study of endothelin receptor blockade in heart failure with diastolic dysfunction and pulmonary hypertension (BADDHY-trial). Heart Lung Circ. 2017;26(5):433–41. https://doi.org/10.1016/j.hlc.2016.09.004.
Vachiery JL, et al. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J. 2018; in press
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Bhavadharini Ramu and Brian A. Houston declare no conflicts of interest.
Ryan J. Tedford reports personal fees from Actelion (Johnson and Johnson) and personal fees from St. Jude Medical (Abbott) outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pharmacologic Therapy
Rights and permissions
About this article

Cite this article
Ramu, B., Houston, B.A. & Tedford, R.J. Pulmonary Vascular Disease: Hemodynamic Assessment and Treatment Selection—Focus on Group II Pulmonary Hypertension. Curr Heart Fail Rep 15, 81–93 (2018). https://doi.org/10.1007/s11897-018-0377-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-018-0377-9