
Abstract
Purpose of review
Over the last years, evidence is accumulating that enhanced late sodium current (INaL) in cardiac pathologies has fundamental consequences for cellular electrophysiology. This review discusses the underlying mechanisms of INaL-induced arrhythmias and the significance of INaL-inhibition as a possible therapeutic approach.
Recent Findings
Inhibition of enhanced INaL, e.g., by ranolazine, was shown to reverse these effects in different myocardial diseases including heart failure. The antianginal drug ranolazine has already been examined in larger clinical trials with promising antiarrhythmic actions.
Summary
Enhanced INaL was found to be present in several cardiac pathologies like ischemia, long QT syndromes, hypertrophic cardiomyopathy, and heart failure. In settings of enhanced INaL, a sodium-dependent calcium overload leads to severe impairment of excitation-contraction coupling and therefore has a high proarrhythmogenic potential. Experimental data showed that inhibition of INaL has a high antiarrhythmic potential which could be confirmed in further clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Heart failure constitutes one of the major causes for morbidity and mortality within the western world. Approximately 1–2% of the population is affected and prevalence dramatically increases after the age of 70. Therefore, heart failure represents a major burden for public health in an aging population [1]. Up to 50% of these heart failure patients die from arrhythmias and sudden cardiac death. However, pharmacological antiarrhythmic therapy of those patients is limited or inefficient [2].
The underlying causes for these arrhythmias are complex structural and electrical remodeling processes in response to myocardial injury. Electrical remodeling has been linked to development of atrial fibrillation and potentially lethal ventricular arrhythmias. Major determinants of electrical remodeling in heart failure involve alteration of numerous ion channels and disturbed intracellular Ca2+ cycling. A major issue of electrical remodeling in heart failure is prolongation of the action potential (AP) with possible occurrence of early afterdepolarizations (EADs).
A complex interplay between different ion currents is involved in remodeling of the cardiac AP. This includes outward K+ currents (Ik), inward Ca2+ currents (ICa), and a persistent component of the inward Na+ currents (INa). Additionally, altered current densities and changes in the spatial distribution of IK, ICa, and INa occur in the presence of heart failure [3]. An important role in AP prolongation and arrhythmias is attributed to the persistent or late component of the inward Na+-current (INaL) [4,5,6,7,8,9].
The Late Sodium Current
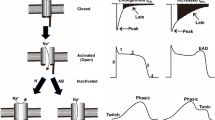
Cardiac excitation depends on highly and well-coordinated voltage-gated sodium channels (NaV) that generate the AP upstroke [10, 11]. NaV are inactivated as quickly as they are activated which is required for cell membrane repolarization and electrical stability. While voltage-dependent inactivation readily switches off most of NaV (INa) current, a small portion of persistent (late) sodium current (INaL) is present even in physiological conditions [4]. A direct link is established between augmented INaL and increased vulnerability for arrhythmias [5]. Many studies have provided evidence that INaL is increased in heart failure which subsequently leads to AP prolongation and arrhythmias [4,5,6,7]. Moreover, INaL is enhanced in several other pathophysiological conditions such as hypertrophy, ischemia, and atrial fibrillation (Fig. 1) [8, 12,13,14,15].
Despite plenty of research has been done on INaL, there still is limited knowledge about the underlying mechanisms of INaL augmentation in cardiac pathologies. As some of the neuronal sodium channels were additionally shown to be expressed in the heart, defining the origin of INaL augmentation even got more complicated [16].

Inward sodium current can be divided into a peak and a late component; both components are also present under physiologic conditions; late sodium current is known to be enhanced in cardiac pathology
Cardiac Sodium Channels
The “cardiac” sodium channel NaV1.5 is encoded by the SCN5A gene and is expressed as the primary cardiac sodium channel in all excitable tissue in the heart. Several different proteins have been identified regulating expression and function of the channel [17]. Malfunctions of NaV1.5 whether congenital or acquired are associated with cardiac disorders and arrhythmias. Mutations in the SCN5A gene have been linked to congenital arrhythmias like long QT syndrome type 3 and Brugada syndrome [18]. Moreover, regulation of the channel is changed under pathological conditions as already mentioned above. Regulatory changes are caused by altered post-translational modifications through associated proteins that modulate biophysical function of the channel [19, 20]. In this case, a major role is attributed to Ca2+/calmodulin-dependent protein kinase II (CaMKII) which is one of the key players in cardiac pathophysiology. The predominant cardiac isoform CaMKIIδ was also described to slow the fast inactivation of inward sodium current [6, 21]. Phosphorylation by CaMKII and other kinases is known to shift voltage dependence of current activation and inactivation as well as a negative shift of channel availability [22, 23]. All these mechanisms could contribute to increased INaL via NaV1.5. Furthermore, different phosphorylation sites for CaMKIIδ and other kinases at NaV1.5 have been identified [24, 25].
NaV1.5 early became a target of antiarrhythmic therapy. Vaughn-Williams divided sodium channel inhibitors in three classes (Ia, Ib, Ic), which differ regarding their effects on action potential duration (APD) and effective refractory period [26]. The CAST Study evaluated the potential of reducing sudden cardiac death by NaV inhibition in patients after myocardial infarction. However, the study was canceled after 10 months because of increased mortality compared to placebo. That is why class I antiarrhythmics and most other antiarrhythmic drugs apart from amiodarone are contraindicated in patients with significant structural heart disease now [27, 28]. Therefore, a major group of patients that require antiarrhythmic treatment have limited therapeutic options. Most importantly, the majority of these patients need longer or lifelong antiarrhythmic treatment with these compounds. This raises the need for novel antiarrhythmic strategies that act more specific, e.g., via INaL and do not affect cardiac conduction by inhibiting the peak sodium current (INa,peak).
Non-cardiac Sodium Channels
Studies showing an association of electrocardiographic (ECG) abnormalities with epilepsy [29] and myotonic disorders [30] lead to the idea that mutated non-cardiac sodium channels might also cause electrophysiological disturbances in the heart. These non-cardiac sodium channel isoforms were later identified in cardiac tissue [16, 31, 32].
Some studies suggested that non-cardiac NaV may contribute to INaL augmentation. A report by Biet et al. showed a significant (∼50%) contribution to INaL by non-cardiac NaV isoforms in healthy canine cardiomyocytes [32]. In another study, Xi et al. proposed that an increased INaL may be explained by overexpression of neuronal NaV1.1 and NaV1.6 in a rat HF model [9]. Another group suggested a relevant contribution of NaV1.1 to INaL in a dog HF model [33].
In recent years, the neuronal sodium channel NaV1.8 was also proposed to contribute to INaL in cardiomyocytes as it was shown to be expressed in mouse hearts [34]. Moreover, expression in intracardiac neurons [35] and human heart tissue was demonstrated [36]. Furthermore, genome-wide association studies (GWAS) have reported that single nucleotide polymorphisms in the SCN10A gene, which encodes NaV1.8, are associated with modulation of cardiac conduction, as well as heart rate and arrhythmic risk [37,38,39].
Interestingly, Yang et al. showed that A-803467, a specific blocker of NaV1.8, can selectively block INaL in rabbit and mouse ventricular cardiomyocytes and, therefore, shortens the APD without any impact on INa,peak [34]. A recent study reported discovery of a novel selective and orally bioavailable NaV1.8 blocker PF-01247324 which modulates augmented INaL in sensory neurons [40]. Moreover, different studies reported that NaV1.8 significantly contributes to INaL triggers and arrhythmogenesis. A recent study showed coding sequence variations in the SCN10A gene to be associated with vulnerability to atrial fibrillation. Electrophysiological studies showed increased INaL for most of the variants [41]. Therefore, novel physiological blockers specifically targeting NaV1.8 may be an interesting therapeutic option for experimental treatment of arrhythmias.
These findings on non-cardiac sodium channels lead to the consideration that inhibition of these channels could provide an approach targeting INaL without affecting INa,peak. by influencing NaV1.5.
Proarrhythmogenic-Enhanced Late Sodium Current
According to current knowledge, augmented INaL is part of an ongoing vicious circle in cardiac pathology. Especially INaL in relation to CaMKII constitutes a key player of cardiac disease [42], which makes it an important issue. CaMKII is known to phosphorylate several ion channels and other proteins involved in excitation-contraction coupling [43]. Furthermore, it was shown to be activated in several pathological conditions of the myocardium [44]. As mentioned before, phosphorylation by CaMKII affects kinetics of the cardiac sodium channel NaV1.5 [21, 24, 25]. This enhances the INaL and therefore increases intracellular sodium ([Na]i). As a consequence, reverse mode of Na+/Ca2+ exchanger (NCX) is activated causing enhanced Ca2+ influx [45]. Elevated intracellular calcium ([Ca2+]i) subsequently activates both ryanodine receptors (RyR2) and CaMKII. CaMKII further phosphorylates RyR2, leading to diastolic Ca2+ leak [46], and NaV1.5, stimulating the cycle again. Elevated diastolic Ca2+ is extruded by forward mode of NCX, generating an inward sodium current again.
Increased inward sodium currents during the action potential plateau and prolonged APD result in early afterdepolarizations (EADs) [47]. Diastolic Ca2+ extrusion via NCX can generate a depolarizing current leading to delayed afterdepolarizations (DADs) [48]. Both EADs and DADs can result in life-threatening arrhythmias (Fig. 2).

Vicious circle of increased late sodium current in cardiac pathology; sodium-dependent calcium overload triggers Ca2+/calmodulin-dependent protein kinase II activity, and enhanced inward sodium current results in action potential prolongations and early afterdepolarizations due to sodium overload; increased diastolic calcium is extruded via NCX causing a depolarizing inward sodium current with potential delayed afterdepolarizations; increased Ca2+/calmodulin-dependent protein kinase II activity actuates the vicious circle again
As CaMKII regulates various proteins in intracellular myocardial signaling, it represents an unspecific target for a therapeutic antiarrhythmic approach. Additionally, by now, there is no clinical substance known to inhibit CaMKII specifically in cardiac tissue. Therefore, inhibiting augmented INaL might constitute the most reasonable target to break the vicious circle.
Therapeutic Inhibition of INaL
As an enhanced INaL was found in several cardiac pathologies, it became an interesting target for pharmacological inhibition. Over the last years, new agents inhibiting INaL were discovered and established drugs were studied for their detailed effects on INaL. Class I antiarrhythmics such as lidocaine and flecainide were shown to have inhibitory effects on INaL, as well as class III antiarrhythmic amiodarone [49]. However, these compounds were not selective enough for INaL compared to INa,peak.
The best examined clinically approved compound inhibiting INaL is ranolazine, which was primarily released as an anti-ischemic drug. Later, ranolazine was found to inhibit INaL potently up to 38-fold higher than INa,peak [50, 51]. Several experimental studies showed ranolazine to reduce [Na]i, thereby NCX reverse mode and diastolic Ca2+ overload in heart failure, ischemia, and oxidative stress [6, 52, 53].
In myocardial trabeculae from human end-stage failing hearts, ranolazine reduced the excessive increase in diastolic tension [54]. In papillary muscles of transgenic CaMKII-overexpressing mice, it also attenuates diastolic dysfunction [55]. Similar results were found by Coppini et al. in isolated ventricular myocytes and trabeculae from patients with hypertrophic cardiomyopathy. Treatment with ranolazine resulted in a faster kinetics of the Ca2+ transients and lower diastolic Ca2+. Both resulting in an accelerated contraction-relaxation cycle and therefore improved diastolic function [8]. Further experimental in vivo studies demonstrated reduced left ventricular end-diastolic pressure and increased left ventricular ejection fraction and stroke volume after acute infusion with ranolazine in a canine heart failure model [56]. A first proof-of-concept study to evaluate the effects of ranolazine in diastolic heart failure was the RALI-DHF study. It showed a significant decrease in left ventricular end-diastolic pressure 30 min after infusion of ranolazine. However, relaxation parameters measured by echocardiography were unchanged [57]. In this context, newer experimental data from Coppini et al. should be mentioned. They showed that INaL inhibition with ranolazine prevented the phenotype development in a mouse model of hypertrophic cardiomyopathy [58].
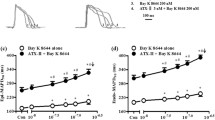
An enhanced INaL is potentially involved in arrhythmogenesis by changing cellular electrophysiology. Therefore, different compounds were tested regarding their potential to selectively inhibit this current. Tetrodotoxin (TTX) is historically known to inhibit the cardiac sodium channel isoform NaV1.5. Maltsev et al. showed that application of 10 μmol/L of TTX not only reversibly blocked INaL but also abbreviate APD and suppressed EADs in cardiomyocytes from human failing hearts [4]. As mentioned before, ranolazine was also found to inhibit INaL in cardiomyocytes, thereby suppressing EADs in a model of long QT syndrome [59]. Further research examined the antiarrhythmic effects of INaL inhibitors in settings of pharmacologically enhanced INaL. A potent inducer of INaL is sea anemone toxin (ATX-II) which consequently induces APD prolongation, EADs, and DADs in different experimental settings [54, 60]. In the light of this, it was demonstrated that ATX-II-induced effects on APD, EADs, and INaL could be reduced by both TTX [4] and ranolazine [59]. Increased CaMKII expression was shown to enhance INaL by direct interaction with the cardiac sodium channel. In a model with transgenic overexpressed CaMKII activity where INaL was enhanced, CaMKII-inhibition could prevent INaL enhancement and in further consequence APD prolongation and arrhythmias [21]. In the same model, later INaL inhibition by ranolazine was also shown to reduce arrhythmias significantly [61]. INaL inhibition by TTX or ranolazine was also shown to act as antiarrhythmic in pathological conditions of enhanced INaL. Our group could demonstrate that ranolazine and TTX normalize enhanced INaL in a model of pressure-induced heart failure. Accordingly, APD was abbreviated by both drugs [6]. Similar results could be observed by Coppini et al. in isolated ventricular cardiomyocytes from patients with hypertrophic cardiomyopathy indicating the significant contribution of INaL to APD prolongation and electrical instability in the failing heart [8].
A second mechanism of INaL contributing to proarrhythmia is the formation of DADs. These arrhythmogenic triggers result from sodium-dependent calcium overload, which is caused by reverse mode of NCX due to elevated Na+ concentration and prolonged APD. This diastolic Ca2+ overload also causes CaMKII activation and thereby as a consequence RyR2-phosphorylation diastolic Ca2+ leak of the sarcoplasmic reticulum also in the human heart [55, 62, 63]. Inhibition of either CaMKII or INaL was shown to reduce diastolic SR-Ca2+ leak and to suppress the occurrence of DADs [6, 62, 64]. As DADs appear to be Ca2+ dependent, Song and coworkers nicely demonstrated the RyR dependence of INaL-induced arrhythmias [48]. They showed ranolazine to prevent APD prolongation and EADs as well as DADs after induction of INaL with ATX-II [48]. Further, they used the sarcoplasmic reticulum Ca2+-release channel inhibitor ryanodine, Ca2+-chelating agents, or the NCX inhibitor KB-R7943 to prevent diastolic Ca2+ overload. After induction of INaL with ATX-II, in the presence of the abovementioned agents, EADs, but no DADs, were observed in this setting. This leads to the suggestion that DADs occur in settings of Ca2+ overload, while formation of EADs is Ca2+ independent. The suppression of DADs by ranolazine was further demonstrated in several conditions with enhanced INaL such as human heart failure [64], pressure-induced heart failure [6], or hypertrophic cardiomyopathy [8].
Based on this promising experimental data, the antiarrhythmic effects of ranolazine were observed in clinical trials. Most information on antiarrhythmic effects of ranolazine were gathered from the MERLIN-TIMI 36 trial. The MERLIN-TIMI 36 trial evaluated ranolazine in patients with non-ST elevation acute coronary syndromes (NSTE-ACS). In contrast to the CAST Study, the incidence of sudden cardiac death was not increased. In fact, there was a numerical reduction of sudden cardiac death close to 45% in patients with a left ventricular ejection fraction <40% where INaL is expected to be enhanced. Moreover, treatment with ranolazine significantly reduced the incidence of non-sustained ventricular tachycardia (more than eight beats) by ∼35% [65]. Elevated levels of the B-type natriuretic peptide (BNP), as it is known for heart failure, are known to be linked with increased risk in ACS patients. Interestingly, in a subgroup of patients from the MERLIN-TIMI 36 trial, who had elevated BNP levels, the combined primary end points out of cardiovascular death, myocardial infarction, and recurrent ischemia were reduced significantly [66]. As mentioned before, ranolazine causes a slight prolongation of the QTc interval. In a retrospective analysis, NSTE-ACS patients with prolonged QTc interval were observed to have an increased risk for sudden cardiac death. At this point, it should be mentioned that treatment with ranolazine was not associated with increased risk for sudden cardiac death compared to placebo in those patients [67]. In other clinical studies, ranolazine caused a modest QTc interval prolongation, whereas in patients with long QT syndrome type 3, QTc interval was shortened [68, 69]. Experimental data showed ranolazine to inhibit INaL with a higher potency than other currents like IKr which would explain shortening of the AP in contrast to prolongation due to IKr under conditions of an enhanced INaL [70].
Besides the MERLIN-TIMI 36 study, other studies also report inhibition of INaL with ranolazine to act antiarrhythmic. Nevertheless, most other studies are case reports or not randomized or placebo controlled. A case series including eight patients suffering from cardiomyopathy reported a 60% reduction of premature ventricular contraction (PVC) burden in six patients with >10% PVCs. In two patients, a PVC-induced cardiomyopathy was supposed, which was normalized after treatment with ranolazine. Additionally, in two patients with sustained ventricular tachycardia, ranolazine terminated the tachycardia and therefore prevented shocks from the implantable cardioverter defibrillator (ICD) [71]. Another study examined patients with ischemic heart disease suffering from antiarrhythmic drug refractory ventricular tachycardia and ICD shocks. Ninety-two percent of the patients had a significant reduction of VTs and no ICD shocks over a follow-up of 6 months under ranolazine medication [72].
Besides the effects of INaL in the ventricle, INaL and its inhibition have also been evaluated in the atria. Atrial fibrillation (AF) is the most common arrhythmia associated with increased rate of morbidity and mortality and is often associated with heart failure. In atrial myocytes from AF patients, INaL was also found to be enhanced, while INa,peak was decreased [15]. However, the formation of arrhythmias in atria is rather complex than in ventricles, and antiarrhythmic properties of ranolazine in atria also include a relevant inhibition of INa,peak [15, 73, 74]. Nevertheless, in atrial myocytes isolated from AF patients, CaMKII-dependent SR-Ca2+ leak and elevated diastolic Ca2+ levels were found [62, 75]. Inhibition of INaL in atrial myocytes was shown to reduce CaMKII activation and SR-Ca2+ leak due to reduced RyR phosphorylation at the CaMKII-specific binding site [62, 75]. Therefore, it is likely that INaL plays a role in atrial fibrillation and that relevant anti-AF effects of ranolazine are attributed to INaL inhibition via reduced proarrhythmogenic diastolic SR-Ca2+ release.
The MERLIN-TIMI 36 study showed ranolazine to significantly reduce supraventricular tachycardia and paroxysmal atrial fibrillation in patients with ACS although the incidence of AF was very low [65, 76]. Later larger trials were started, investigating the efficiency and safety of ranolazine alone (RAFFAELLO) or in combination with low-dose dronedarone (HARMONY) for the treatment of paroxysmal atrial fibrillation. Treatment with ranolazine was shown to be safe but ranolazine alone did not significantly reduce recurrence of AF significantly, although pooled data from the 500 and 750 mg groups were close to significance (p = 0.051) [77]. Nevertheless, as it was a small phase 2 study, this data shows a promising potential of ranolazine in atrial fibrillation which needs to be confirmed in further specifically designed trials. A combination of ranolazine and dronedarone reduced the AF burden up to 70% in patients with paroxysmal AF in the HARMONY trial [78].
Another drug, GS-458967, has been introduced recently as a selective INaL blocker [79, 80]. In contrast to ranolazine, GS-458967 has more selective effects on INaL-mediated parameters. Surprisingly, atrial cells were more sensitive to INaL inhibition by GS-458967 than ventricles. However, in ventricles, GS-458967 causes abbreviation of APD during long QT conditions only, suggesting its pathology-specific effects [81]. Nevertheless, GS-458967 reduced ventricular depolarization and repolarization heterogeneity during acute myocardial ischemia in a porcine model [82]. Administration of GS-458967 provided protection against catecholamine-induced ventricular tachycardia and T wave alternans [83]. GS-458967 did not cause alteration in PR and QT intervals or QRS duration as well as in heart rate and arterial blood pressure [82, 83]. One other group demonstrated suppressive effects of GS-458967 on aconitine-induced ventricular tachycardia and fibrillation in rat hearts [84]. However, by now, there is no clinical data for this drug.
A very recent discovery of a potent INaL inhibitor is the drug eleclazine (GS-6615). Its selectivity has been improved over ranolazine and showed 42 times more potency than ranolazine with EC50 8000 nM [85]. In contrast to ranolazine, eleclazine reduces potently INaL but has no major alterations of other ion currents such as ICaL,IKr, IKs, and peak INa [86]. Of note, eleclazine is proved to be superior over flecainide also in suppressing ventricular tachycardia and T wave alternans in a catecholamine-induced porcine model [87].
A recent phase 1 trial showed eleclazine to be safe in patients with type 3 long QT syndrome. Phase 2 and 3 trials to treat long QT-3 syndrome, hypertrophic cardiomyopathy (LIBERTY-HCM), and ventricular arrhythmias in patients with implanted ICDs (TEMPO) were started [88]. Recently, it was reported that the recruitment of the TEMPO trial was stopped because it failed effectivity. Later, LIBERTY-HCM and the trial for long QT-3 syndrome were also stopped [89]. However, to draw distinct conclusions from these trials, the final results have to be published.
Conclusion
An enhanced INaL has been described to play a crucial role for cellular electrophysiology in several cardiac pathologies such as heart failure. Promising experimental data could show that inhibition of an enhanced INaL has the potential to suppress arrhythmias in vitro and in vivo.
Ranolazine as a clinically approved drug for the treatment of ischemia has the potential for dual suppression of atrial and ventricular arrhythmias. This has been demonstrated also in some clinical studies. Nevertheless, future work should concentrate on prospective randomized trials, and more specific designed studies are necessary to prove a significant role of INaL inhibition in clinical antiarrhythmic treatment.
References
Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–46.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37:2129–200.
Cutler MJ, Jeyaraj D, Rosenbaum DS. Cardiac electrical remodeling in health and disease. Trends Pharmacol Sci. 2011;32:174–80.
Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52.
Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res. 2013;99:600–11.
Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–22.
Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83.
Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–84.
Xi Y, Wu G, Yang L, Han K, Du Y, Wang T, et al. Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model. Eur J Heart Fail. 2009;11:749–57.
Marionneau C, Abriel H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J Mol Cell Cardiol. 2015;82:36–47.
Hund TJ, Mohler PJ. Nav channel complex heterogeneity: new targets for the treatment of arrhythmia? Circulation. 2014;130:132–4.
Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–41.
Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. 1996:337–47.
Huang B, El-Sherif T, Gidh-Jain M, Qin D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol. 2001;12:218–25.
Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–42.
Maier SKG, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–8.
Rook MB, Evers MM, Vos MA, Bierhuizen MFA. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12–23.
Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–48.
Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431–45.
Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886–94.
Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38.
Ono K, Fozzard HA, Hanck DA. Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ Res. 1993;72:807–15.
Schubert B, VanDongen AM, Kirsch GE, Brown AM. Beta-adrenergic inhibition of cardiac sodium channels by dual G-protein pathways. Science. 1989;245:516–9.
Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, et al. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–77.
Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–69.
Vaughan Williams EM. Classifying antiarrhythmic actions: by facts or speculation. J Clin Pharmacol. 1992;32:964–77.
Gintant GA, Gallacher DJ, Pugsley MK. The “overly-sensitive” heart: sodium channel block and QRS interval prolongation. Br J Pharmacol. 2011;164:254–9.
Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. N Engl J Med. 1991;324:781–8.
Akalin F, Tirtir A, Yilmaz Y. Increased QT dispersion in epileptic children. Acta Paediatr. 2003;92:916–20.
Komajda M, Frank R, Vedel J, Fontaine G, Petitot JC, Grosgogeat Y. Intracardiac conduction defects in dystrophia myotonica. Electrophysiological study of 12 cases. Br Heart J. 1980;43:315–20.
Haufe V, Camacho JA, Dumaine R, Günther B, Bollensdorff C, von Banchet GS, et al. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol. 2005;564:683–96.
Biet M, Barajas-Martínez H, Ton A-T, Delabre J-F, Morin N, Dumaine R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J Mol Cell Cardiol. 2012;53:593–8.
Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Nav 1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol. 2015;593:1409–27.
Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–32.
Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111:333–43.
Chambers JC, Zhao J, Terracciano CMN, Bezzina CR, Zhang W, Kaba R, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–52.
Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–22.
Sotoodehnia N, Isaacs A, de Bakker PIW, Dörr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–76.
Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–85.
Payne CE, Brown AR, Theile JW, Loucif AJC, Alexandrou AJ, Fuller MD, et al. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol. 2015;172:2654–70.
Savio-Galimberti E, Weeke P, Muhammad R, Blair M, Ansari S, Short L, et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res. 2014;104:355–63.
Fischer TH, Maier LS, Sossalla S. The ryanodine receptor leak: how a tattered receptor plunges the failing heart into crisis. Heart Fail Rev. 2013;18:475–83.
Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–7.
Maier LS, Bers DM, Brown JH. Calmodulin and Ca2+/calmodulin kinases in the heart—physiology and pathophysiology. Cardiovasc Res. 2007;73:629–30.
Pieske B, Maier LS, Piacentino V, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–53.
Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–11.
January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–90.
Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. AJP Hear Circ Physiol. 2008;294:H2031–9.
Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart BMJ Group. 2006;92(Suppl 4):iv6–iv14.
Sossalla S, Maier LS. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther. 2012;133:311–23.
Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S169–77.
Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–22.
Zhang XQ, Yamada S, Barry WH. Ranolazine inhibits an oxidative stress-induced increase in myocyte sodium and calcium loading during simulated-demand ischemia. J Cardiovasc Pharmacol. 2008;51:443–9.
Sossalla S, Wagner S, Rasenack ECL, Ruff H, Weber SL, Schöndube FA, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43.
Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–61.
Sabbah HN, Chandler MP, Mishima T, Suzuki G, Chaudhry P, Nass O, et al. Ranolazine, a partial fatty acid oxidation (pFOX) inhibitor, improves left ventricular function in dogs with chronic heart failure. J Card Fail. 2002;8:416–22.
Maier LS, Layug B, Karwatowska-Prokopczuk E, Belardinelli L, Lee S, Sander J, et al. RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction. JACC Hear Fail. 2013;1:115–22.
Coppini R, Mazzoni L, Ferrantini C, Gentile F, Pioner JM, Laurino T, et al. Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ Hear Fail. 2017;10:e003565. CLINICAL PERSPECTIVE
Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605.
Boutjdir M, el Sherif N. Pharmacological evaluation of early afterdepolarisations induced by sea anemone toxin (ATXII) in dog heart. Cardiovasc Res. 1991;25:815–9.
Sossalla S, Maurer U, Schotola H, Hartmann N, Didié M, Zimmermann W-H, et al. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIδC can be reversed by inhibition of late Na+ current. Basic Res Cardiol. 2011;106:263–72.
Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res. 2015;107:184–96.
Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, et al. Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation. 2013;128:970–81.
Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J Mol Cell Cardiol. 2014;76:94–105.
Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non-ST-segment-elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–52.
Morrow DA, Scirica BM, Sabatine MS, de Lemos JA, Murphy SA, Jarolim P, et al. B-Type natriuretic peptide and the effect of ranolazine in patients with non–ST-segment elevation acute coronary syndromes. J Am Coll Cardiol. 2010;55:1189–96.
Karwatowska-Prokopczuk E, Wang W, Cheng ML, Zeng D, Schwartz PJ, Belardinelli L. The risk of sudden cardiac death in patients with non-ST elevation acute coronary syndrome and prolonged QTc interval: effect of ranolazine. Europace. 2013;15:429–36.
Wilson SR, Scirica BM, Braunwald E, Murphy SA, Karwatowska-Prokopczuk E, Buros JL, et al. Efficacy of ranolazine in patients with chronic angina observations from the randomized, double-blind, placebo-controlled MERLIN-TIMI (Metabolic Efficiency With Ranolazine for Less Ischemia in Non-ST-Segment Elevation Acute Coronary Syndromes) 36 Trial. J Am Coll Cardiol. 2009;53:1510–6.
Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–93.
Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–10.
Yeung E, Krantz MJ, Schuller JL, Dale RA, Haigney MC. Ranolazine for the suppression of ventricular arrhythmia: a case series. Ann Noninvasive Electrocardiol. 2014;19:345–50.
Bunch TJ, Mahapatra S, Murdock D, Molden J, Weiss JP, May HT, et al. Ranolazine reduces ventricular tachycardia burden and ICD shocks in patients with drug-refractory ICD shocks. Pacing Clin Electrophysiol. 2011;34:1600–6.
Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–7.
Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–57.
Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–44.
Scirica BM, Belardinelli L, Chaitman BR, Waks JW, Volo S, Karwatowska-Prokopczuk E, et al. Effect of ranolazine on atrial fibrillation in patients with non-ST elevation acute coronary syndromes: observations from the MERLIN-TIMI 36 trial. Europace. 2015;17:32–7.
De Ferrari GM, Maier LS, Mont L, Schwartz PJ, Simonis G, Leschke M, et al. Ranolazine in the treatment of atrial fibrillation: results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following An ELectricaL CardiOversion) study. Hear. Rhythm. 2015;12:872–8.
Reiffel JA, Camm AJ, Belardinelli L, Zeng D, Karwatowska-Prokopczuk E, Olmsted A, et al. The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ Arrhythm Electrophysiol. 2015;8:1048–56.
Belardinelli L, Liu G, Smith-Maxwell C, Wang W-Q, El-Bizri N, Hirakawa R, et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32.
Sicouri S, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Hear. Rhythm. 2013;10:1036–43.
Burashnikov A, Di Diego JM, Goodrow RJ, Belardinelli L, Antzelevitch C. Atria are more sensitive than ventricles to GS-458967-induced inhibition of late sodium current. J Cardiovasc Pharmacol Ther. 2015;20:501–8.
Bonatti R, Silva AFG, Batatinha JAP, Sobrado LF, Machado AD, Varone BB, et al. Selective late sodium current blockade with GS-458967 markedly reduces ischemia-induced atrial and ventricular repolarization alternans and ECG heterogeneity. Hear. Rhythm. 2014;11:1827–35.
Alves Bento AS, Bacic D, Saran Carneiro J, Nearing BD, Fuller H, Justo FA, et al. Selective late INa inhibition by GS-458967 exerts parallel suppression of catecholamine-induced hemodynamically significant ventricular tachycardia and T-wave alternans in an intact porcine model. Hear. Rhythm. 2015;12:2508–14.
Pezhouman A, Madahian S, Stepanyan H, Ghukasyan H, Qu Z, Belardinelli L, et al. Selective inhibition of late sodium current suppresses ventricular tachycardia and fibrillation in intact rat hearts. Hear. Rhythm. 2014;11:492–501.
Zablocki JA, Elzein E, Li X, Koltun DO, Parkhill EQ, Kobayashi T, et al. Discovery of dihydrobenzoxazepinone (GS-6615) late sodium current inhibitor (late INai), a phase II agent with demonstrated preclinical anti-ischemic and antiarrhythmic properties. J Med Chem. 2016;59:9005–17.
Rajamani S, Liu G, El-Bizri N, Guo D, Li C, Chen X-L, et al. The novel late Na(+) current inhibitor, GS-6615 (eleclazine) and its anti-arrhythmic effects in rabbit isolated heart preparations. Br J Pharmacol. 2016;173:3088–98.
Bacic D, Carneiro JS, Bento AA, Nearing BD, Rajamani S, Belardinelli L, et al. Eleclazine, an inhibitor of the cardiac late sodium current, is superior to flecainide in suppressing catecholamine-induced ventricular tachycardia and T-wave alternans in an intact porcine model. Hear Rhythm. 2017;14:448–54.
Olivotto I, Hellawell JL, Farzaneh-Far R, Blair C, Coppini R, Myers J, et al. Novel approach targeting the complex pathophysiology of hypertrophic cardiomyopathy: the impact of late sodium current inhibition on exercise capacity in subjects with symptomatic hypertrophic cardiomyopathy (LIBERTY-HCM) trial. Circ Heart Fail. 2016;9:e002764.
End of the road for eleclazine and liberty HCM study—HCM beat [Internet]. [cited 2017 Mar 31]. Available from: https://hcmbeat.com/2016/12/27/end-of-the-road-for-eleclazine-and-liberty-hcm-study/
Acknowledgements
PB is supported by a clinical researcher grant by the College of Translational Medicine of the Ministry for Science and Culture, State of Lower Saxony; SA is funded by the Marga and Walter Boll Foundation; STS is also funded by the Marga and Walter Boll Foundation and the German Center for Cardiovascular Research (DZHK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Philipp Bengel and Shakil Ahmad each declare no potential conflicts of interest.
Samuel Sossalla receives speaker’s honoraria from Berlin-Chemie & Menarini (provider of ranolazine).
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Experimental Therapeutics
Rights and permissions
About this article

Cite this article
Bengel, P., Ahmad, S. & Sossalla, S. Inhibition of Late Sodium Current as an Innovative Antiarrhythmic Strategy. Curr Heart Fail Rep 14, 179–186 (2017). https://doi.org/10.1007/s11897-017-0333-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-017-0333-0