
Abstract
Heart failure in the setting of chronic kidney disease (CKD) is an increasingly common scenario and carries a poor prognosis. Clinicians lack tools for primary or secondary heart failure prevention in patients with cardiorenal syndromes. In patients without CKD, angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin receptor blockers (ARB) and statins mitigate cardiovascular risk in large part due to salutary effects on the endothelium. In the setting of CKD, use of these therapies is limited by adverse effects of hyperkalemia in pre-dialysis CKD (ACE-I/ARB), or potential increased risk of stroke in end-stage renal disease (statins). The soluble guanylate cyclase (sGC) stimulators are a novel class of medications that promote endothelial and myocardial function with no known risk of hyperkalemia or stroke. In this review, we discuss the evidence emerging from recent clinical trials of sGC stimulators in pulmonary hypertension and heart failure, the diseased pathways involved in cardiorenal syndromes likely to be restored by sGC stimulators, and several strategies for designing future clinical trials of cardiorenal syndromes that might shorten the timeline for discovery and approval of effective cardiovascular therapies in these high-risk patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is the most common cardiovascular event [1] among the 20 million Americans with chronic kidney disease (CKD) (defined as an estimated glomerular filtration rate [eGFR] <60 ml/min/m2 or albuminuria) [2]. Several investigations have focused on the prevalence and ramifications of reduced eGFR or albuminuria in patients with HF [3], as well as the risks of hyperkalemia and worsening renal function associated with inhibition of the renin angiotensin aldosterone system (RAAS) in these patients [4]. Many of these patients have some form of cardiorenal syndrome, whether type 4 (CKD leading to cardiovascular disease [CVD]) or type 2 (chronic CVD leading to CKD) [5]. In 2008, approximately 10 % of the $200 billion United States Medicare budget was devoted to patients with concomitant CKD and CVD, highlighting the significant economic burden of cardiorenal syndromes [6].
Despite the public health impact of co-existing HF and renal disease, we lack safe and effective preventive or therapeutic options for cardiorenal syndromes, particularly in patients who already have CKD. Contributing factors to this treatment gap include an incomplete understanding of the mechanisms of CVD in patients with advanced CKD; the risks of hyperkalemia and worsening eGFR associated with the use of angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin receptor blockers (ARB) in advanced CKD; exclusion of patients with CKD from most HF trials due to safety issues or high burden of comorbidities; non-specific inclusion criteria for cardiovascular trials in CKD; and difficulty recruiting and retaining patients with advanced CKD into clinical trials.
The sGC stimulators are a novel class of medications that activate the enzymatic activity of sGC to produce cyclic guanosine monophosphate (cGMP), an important mediator of endothelial function. The endogenous stimulator of sGC, nitric oxide (NO), is generated by vascular endothelium, and diffuses into adjacent target tissues. sGC itself, the “NO receptor” is located intracellularly in all tissues, with highest expression levels in the myocardium. When NO is inactivated or low, sGC stimulation by these new agents occurs directly in target tissues. Importantly, the sGC stimulators could ameliorate endothelial dysfunction without the risk of hyperkalemia, which prevents the use of ACE-I/ARB in up to one third of CKD patients [7]. Here, we review the pathophysiology of the cGMP pathway, discuss the potential advantages of sGC stimulators for patients with HF and cardiorenal syndromes, and propose strategies for efficient trial design of this novel therapy.
Pathways Leading to HF in Patients with Cardiorenal Syndromes
There are several known pathways leading to HF in patients with cardiorenal syndromes. Left ventricular hypertrophy is prevalent in over 50 % of CKD patients; lower eGFR is associated with higher left ventricular mass across all stages of CKD [8]. The elegant work of Wolf and colleagues has identified the phosphaturic hormone fibroblast growth factor (FGF)-23 as a kidney-specific mediator of left ventricular hypertrophy [9] and predictor of HF in CKD [10]. Anemia is an important risk factor, and iron deficiency may be an important link between anemia, bone metabolism, and cardiovascular disease in CKD [11]. At all stages of CKD, deranged mineral metabolism contributes to vascular calcification, leading to accelerated atherosclerotic disease characterized by medial and intimal plaque calcification. Renin-aldosterone activation leads to sympathetic overactivity, volume overload, and cardiac remodeling in patients with CKD.
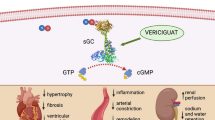
There are at least two additional aspects of CKD that, while less studied, implicate cGMP deficiency as a modifiable, pathogenic factor in the development of HF in this population. First, endothelial dysfunction is a characteristic of CKD, and lower eGFR is associated with severity endothelial dysfunction across stages of CKD 1–5 [12]. Mechanisms for endothelial dysfunction in CKD include oxidative stress and higher levels of asymmetric dimethylarginine [12], both of which reduce NO bioavailability. NO is the first signal in the NO-cGMP pathway leading up to intracellular cGMP production, which in turn results in beneficial endothelial properties including vasorelaxation, anti-proliferation, anti-platelet, anti-inflammation, and anti-fibrosis (Fig. 1) [13, 14]. Second, right ventricle (RV) dysfunction and pulmonary hypertension are under- appreciated aspects of cardiorenal syndromes. Pulmonary hypertension is common in end-stage renal disease (ESRD) [15]. A recent study extended these findings to study participants with pre-dialysis CKD, in whom 23 % had estimated pulmonary artery systolic pressure (ePASP) >35 mmHg, and ePASP was associated with adverse cardiovascular outcomes independently of eGFR and left ventricular hypertrophy [16]. Pulmonary hypertension in CKD may be due to increased preload from arteriovenous fistula, abnormal LV diastolic function, obstructive sleep apnea, endothelial function causing pulmonary vasoconstriction, or increased pulmonary vascular stiffening [17]. A therapy such as the sGC stimulators that upregulate production of cGMP independently of NO might overcome NO deficiency in CKD and improve pulmonary endothelial function, with subsequent improvement in pulmonary hypertension (pulmonary vascular resistance, pulmonary artery compliance, and right ventricular-pulmonary artery coupling) in these patients.

Actions of the cGMP stimulators on systems involved in cardiorenal syndromes. Starting at the top left, this figure illustrates the production of cGMP within the smooth muscle cell via endogenous NO from the endothelium or by exogenous sGC stimulators/activators. The sGC α/β heterodimer is pictured in the native, heme(+) form and the oxidized, heme(−) form. sGC soluble guanylate cyclase, eNOS endothelial nitric oxide synthase, GTP guanosine triphosphate, cGMP cyclic guanosine monophosphate
Mainstays of Primary and Secondary HF Prevention Have Adverse Effects in CKD: ACE-I/ARB, Statins
The need for novel HF therapies for patients with CKD is compounded by the problems associated with the use of ACE-I/ARB in this population. Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend these medications in patients with eGFR <60 ml/min/1.73 m2 or albuminuria >30 mg per 24 h [18].
However, ACE-I/ARB are often avoided in patients with eGFR < 30 ml/min/1.73 m2 not yet on dialysis, due to the risk of hyperkalemia or worsening eGFR. In a study of renal clinic patients in the Veterans Administration Health System, nearly one third of 279 CKD patients were not on RAAS blockade despite a clinical indication [7]. The problem of hyperkalemia associated with RAAS inhibition in patients with CKD is so widespread that it was recently addressed by a clinical trial of patiromer, a potassium-lowering medication [19]. There may be a lower risk of hyperkalemia with aldosterone antagonists in anuric ESRD patients. Spironolactone was shown to reduce cardiac events in ESRD patients with urine output <200 ml/day; however, there were discontinuations of the drug due to hyperkalemia and other side effects [20]. Whether ACE-I alone benefits patients with ESRD is uncertain [21], although adding an ARB to background ACE-I treatment did improve outcomes for patient on hemodialysis with reduced ejection fraction [22]. As of 2011, 55 % of patients on dialysis with a diagnosis of HF (ejection fraction [EF] not specified) were not on ACE-I or ARB [23]. Furthermore, statins—which form the mainstay of primary and secondary prevention of cardiovascular disease—may be ineffective [24] or associated with increased risk of stroke in ESRD [25].
Evidence Base for the Role of the cGMP Pathway in Pulmonary, Cardiac, and Renal Disease
The diverse functions of the endothelium, including vasorelaxation, anti-proliferation, anti-platelet, anti-inflammation, and anti-fibrosis, depend on an intact NO-cGMP pathway (Fig. 1) [13, 14]. NO diffuses into vascular smooth muscle cells and other tissues to stimulate sGC, which converts guanosine triphosphate (GTP) to cGMP, which in turn leads to production of protein kinase-G (PKG) and PKG’s downstream effects [13].
Cell culture and animal studies have shed light on organ-specific effects of the NO-cGMP pathway. In the lungs, disruption of the NO-cGMP pathway results in pulmonary vascular remodeling, upregulated smooth muscle tone, and platelet aggregation characteristic of pulmonary hypertension [14]. In the heart, modulation of the NO-cGMP pathway within the cardiomyocyte directly influences ventricular distensibility [26]. Phosphorylation of troponin I by cGMP-dependent kinases prevents troponin cross-linking and myocardial stiffening [27]. Animal studies have shown that titin, a key determinant of diastolic cell length and cardiomyocyte length-tension relationship, is also phosphorylated by cGMP-dependent kinases [28]. When the cGMP-PKG pathway is deficient, titin is hypophosphorylated, leading to cardiomyocyte stiffening. In the coronary vascular smooth muscle, the NO-cGMP pathway permits coronary vasorelaxation, which results in improved perfusion and contractility [29].
In the kidney, impairment of the NO-cGMP pathway leads to interstitial fibrosis and renal tubular apoptosis [30, 31]. NO depletion results in dysregulation of afferent and efferent arterioles and impairs renal medullary flow [14]. A recent review enumerates the large body of preclinical evidence that supports the use of sGC stimulators and activators to mitigate these pro-fibrotic and hemodynamic effects of low NO bioavailability [32]. In rats with sub-total nephrectomy, treatment with the sGC activator cinaciguat slowed progression of kidney disease and left ventricular hypertrophy [33]. Rats with anti-thy-1-induced glomerulosclerosis had reduced glomerular and interstitial inflammation, glomerular proliferation, and matrix deposition, after treatment with the sGC stimulator BAY 41–2272 [34]. In low-renin, high-renin, or salt-sensitive hypertensive rats, the sGC stimulator riociguat reduced cardiac and renal fibrosis and improved cardiac and renal function [35, 36].
Given the importance of the NO-cGMP pathway in the lungs, heart, and kidneys, therapies affecting this pathway may have high potential for preventing or treating HF associated with cardiorenal syndrome [14]. In advanced CKD, NO production is severely impaired by high levels of asymmetric dimethylarginine and oxidative stress [37]. High oxidative stress negatively impacts the NO-cGMP pathway at several levels. The oxidant peroxynitrite uncouples endothelial NO synthase (eNOS); the uncoupled enzyme shifts from NO to superoxide production, which in turn generates more peroxynitrite. Downstream of NO production, oxidants deactivate NO, and oxidize sGC to its heme-free state, which is less responsive to NO [13]. sGC stimulators sensitize sGC to NO, upregulate sGC production of cGMP independently of NO, and promote the non-oxidized, more active form of sGC by stabilizing the nitrosyl-heme complex [38]. Because the sGC stimulators bypass NO by acting downstream of NO directly on sGC, these agents are potential candidates for treating endothelial dysfunction in severe CKD. Thus, they are effective even in states of low NO bioavailability—in theory, this gives them an advantage over phosphodiesterase type 5 inhibitors, medications that prevent cGMP degradation but have little impact when NO and cGMP levels are low [13].
Clinical Trials of sGC Stimulators
Short- and long-term effects of the sGC stimulator, riociguat, were studied in patients with pulmonary arterial hypertension (Pulmonary Arterial Hypertension sGC- Stimulator Trial [PATENT]-1 and -2) [39, 40] and chronic thromboembolic pulmonary hypertension (Chronic Thromboembolic Pulmonary Hypertension sGC Stimulator Trial [CHEST]-1 and -2) [41, 42]. Six-minute walk distance and World Health Organization functional class improved over a 6-week time period in patients receiving riociguat, and these improvements were sustained after 1 year.
In the Acute Hemodynamic Effects of Riociguat in Patients with Pulmonary Hypertension Associated with Diastolic Heart Failure (DILATE-1) trial, a single 0.5–2 mg dose of riociguat was given to patients with pulmonary hypertension and heart failure with preserved ejection fraction (HFpEF). While the primary endpoint (reduction in mean pulmonary artery pressure) was not achieved, riociguat did result in higher stroke volume, lower right ventricular end-diastolic volume, and a decrease in systolic blood pressure by 12 mmHg [43]. In the Left Ventricular Systolic Dysfunction Associated with Pulmonary Hypertension Riociguat Trial (LEPHT), 200 patients with pulmonary hypertension due to heart failure with reduced ejection fraction (HFrEF) already on optimal therapy were randomized to placebo or target doses of riociguat 0.5, 1, or 2 mg three times daily over 16 weeks. As in DILATE-1, the primary endpoint, reduction in mean pulmonary artery pressure, was not achieved; however, patients treated in the 2 mg target dose arm had reduced pulmonary and systemic vascular resistance, increased cardiac index, and improved quality of life, without reduction of systolic blood pressure [44]. Notably, DILATE-1 [43] and LEPHT [44] were the only studies to include patients with reduced renal function, and none included patients with eGFR < 30 ml/min/1.73 m2 (Table 1). The parallel-group Soluble Guanylate Cyclase Stimulator in Heart Failure Studies (SOCRATES) trials aimed to study the effects of a once-daily sGC stimulator, vericiguat, in patients recently hospitalized for HFrEF (SOCRATES-Reduced) or HFpEF (SOCRATES-Preserved) [45]. In SOCRATES-Reduced, vericiguat was safe and well-tolerated [46]. The study did not meet its primary endpoint (12-week change in N-terminal pro-B-type natriuretic peptide [NT-proBNP] in the pooled three highest vericiguat dose groups). However, in secondary analyses, there was a dose–response relationship such that NT-proBNP decreased in a step-wise fashion with increasingly higher doses of vericiguat. The SOCRATES-Preserved trial is complete, but the results have not yet been presented. Both SOCRATES studies included patients with moderate CKD (30–59 ml/min/1.73 m2) and should yield important information on safety and efficacy of sGC stimulation in CKD patients who have concomitant HF. However, it is important to note that both SOCRATES trials involved patients recently hospitalized with worsening HF, a time during which renal function may be quite variable. Thus, determining the true severity of CKD in the enrolled patients may be difficult. In addition, patients with worsening chronic HF (such as those enrolled in the SOCRATES trials) have been notoriously difficult to treat in clinical trials. Thus, in terms of the utility of sGC stimulators in cardiorenal syndromes associated with HF, while the SOCRATES trials will provide some insight, future outpatients studies of cardiorenal patients will likely be able to better inform the long-term benefit of sGC stimulators in this patient population.
Design Considerations for a Trial of sGC Stimulators in Patients with HF and Cardiorenal Syndromes
More efficient testing of HF therapies in patients with cardiorenal syndromes could be accomplished by focusing phase II clinical trials on specific subgroups of patients of similar phenotype, and choosing intermediate endpoints that are most relevant to the disease phenotype (Table 2). For example, in HF patients with mildly reduced eGFR, stabilizing renal function is an important long-term goal. In cardiorenal syndromes, an sGC stimulator could preserve renal function by improving cardiac function; alternatively, sGC stimulators could slow progression of renal disease by direct effects on the kidney. Change in eGFR, endothelial function or left ventricular mass would be appropriate intermediate physiological endpoints. In patients with CKD who do not have HF, in whom preventing HFpEF is an important long-term goal, improving LV diastolic function might be an appropriate intermediate endpoint. sGC stimulators could potentially increase cGMP levels, reduce troponin and titin cross-linking and prevent the ventricular stiffening [27, 28, 47] that can lead to HFpEF. Diastolic function, measured by Doppler and tissue Doppler echocardiography, cardiac mechanics measured by speckle-tracking analysis, and left ventricular mass measured by echocardiography or cardiac magnetic resonance (CMR) imaging would be useful intermediate endpoints.
In addition to focusing trials on subgroups of participants with similar cardiac phenotype, a potential strategy for augmenting statistical power, lowering sample size, and shortening trial duration would be to use imaging modalities such as speckle-tracking echocardiography (STE) or CMR imaging. These imaging modalities provide highly reproducible measurements, allowing for a smaller sample size in a clinical trial without sacrificing the power needed to detect a significant change in a given index of cardiac function. STE is sensitive to small decrements of systolic dysfunction among patients with normal EF, and these differences in systolic function predict mortality in patients HFpEF [48] or ESRD [49, 50], providing further support of the utility of STE in this population. CMR provides higher accuracy and reproducibility in measurement of LV mass than either 2D or 3D echocardiography, and unlike cardiac CT, does not require potentially harmful contrast. For example, the sample size needed to detect a 10-g change in LV mass by CMR is only ten patients each in placebo and treatment group; the sample size would need to be six times larger if 2D echo is used instead [51].
A larger sample size, coupled with examination of LV mass by CMR, would allow detection of smaller changes in LV mass, potentially shortening the duration of the clinical trial.
Conclusions
The sGC stimulators may have a particular role in preventing or treating HF in the setting of advanced CKD as they would not be expected to carry the risks of hyperkalemia or worsening renal function that limit the use of ACE-I/ARB in these patients. Focusing future clinical trials on subgroups most likely to benefit and employing sensitive imaging modalities such as STE and cardiac MRI as intermediate physiological endpoints could expedite the evaluation of sGC stimulators and other novel therapies for treatment and prevention of HF associated with cardiorenal syndromes.
References
Cardiovascular disease in patients with CKD, Vol.1 Ch. 4 [http://www.usrds.org/2014/view/v1_04.aspx]
Adult population of the United States [http://www.census.gov].
Damman K, Masson S, Hillege HL, Maggioni AP, Voors AA, Opasich C, et al. Clinical outcome of renal tubular damage in chronic heart failure. Eur Heart J. 2011;32(21):2705–12.
Rossignol P, Dobre D, McMurray JJ, Swedberg K, Krum H, van Veldhuisen DJ, et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail. 2014;7(1):51–8.
Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31(6):703–11.
Costs of chronic kidney disease [http://www.usrds.org/2010/view/v1_09.asp]
Yildirim T, Arici M, Piskinpasa S, Aybal-Kutlugun A, Yilmaz R, Altun B, et al. Major barriers against renin-angiotensin-aldosterone system blocker use in chronic kidney disease stages 3–5 in clinical practice: a safety concern? Ren Fail. 2012;34(9):1095–9.
Park M, Hsu CY, Li Y, Mishra RK, Keane M, Rosas SE, et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol. 2012;23(10):1725–34.
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–408.
Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol. 2014;25(2):349–60.
Charytan DM, Fishbane S, Malyszko J, McCullough PA, Goldsmith D. Cardiorenal syndrome and the role of the bone-mineral axis and anemia. Am J Kidney Dis 2015.
Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T, et al. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am J Kidney Dis. 2006;47(1):42–50.
Stasch JP, Evgenov OV. Soluble guanylate cyclase stimulators in pulmonary hypertension. Handb Exp Pharmacol. 2013;218:279–313.
Gheorghiade M, Marti CN, Sabbah HN, Roessig L, Greene SJ, Bohm M, et al. Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev. 2013;18(2):123–34.
Bolignano D, Rastelli S, Agarwal R, Fliser D, Massy Z, Ortiz A, et al. Pulmonary hypertension in CKD. Am J Kidney Dis. 2013;61(4):612–22.
Bolignano D, Lennartz S, Leonardis D, D’Arrigo G, Tripepi R, Emrich IE, et al. High estimated pulmonary artery systolic pressure predicts adverse cardiovascular outcomes in stage 2–4 chronic kidney disease. Kidney Int. 2015;88(1):130–6.
Di Lullo L, Floccari F, Rivera R, Barbera V, Granata A, Otranto G, et al. Pulmonary hypertension and right heart failure in chronic kidney disease: new challenge for 21st-century cardionephrologists. Cardiorenal Med. 2013;3(2):96–103.
Group. KDIGOKBPW: KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int Suppl. 2012(2):337–414.
Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015;372(3):211–21.
Matsumoto Y, Mori Y, Kageyama S, Arihara K, Sugiyama T, Ohmura H, et al. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J Am Coll Cardiol. 2014;63(6):528–36.
Zannad F, Kessler M, Lehert P, Grunfeld JP, Thuilliez C, Leizorovicz A, et al. Prevention of cardiovascular events in end-stage renal disease: results of a randomized trial of fosinopril and implications for future studies. Kidney Int. 2006;70(7):1318–24.
Cice G, Di Benedetto A, D’Isa S, D’Andrea A, Marcelli D, Gatti E, et al. Effects of telmisartan added to angiotensin-converting enzyme inhibitors on mortality and morbidity in hemodialysis patients with chronic heart failure a double-blind, placebo-controlled trial. J Am Coll Cardiol. 2010;56(21):1701–8.
Cardiovascular Disease Vol 2. Ch. 4 [http://www.usrds.org/atlas.aspx]
Fellstrom BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360(14):1395–407.
Wanner C, Krane V, Marz W, Olschewski M, Mann JF, Ruf G, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353(3):238–48.
Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89(5):2070–8.
Paulus WJ, Bronzwaer JG. Nitric oxide’s role in the heart: control of beating or breathing? Am J Physiol Heart Circ Physiol. 2004;287(1):H8–13.
Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104(1):87–94.
Zeiher AM, Krause T, Schachinger V, Minners J, Moser E. Impaired endothelium-dependent vasodilation of coronary resistance vessels is associated with exercise-induced myocardial ischemia. Circulation. 1995;91(9):2345–52.
Ito K, Chen J, Seshan SV, Khodadadian JJ, Gallagher R, El Chaar M, et al. Dietary arginine supplementation attenuates renal damage after relief of unilateral ureteral obstruction in rats. Kidney Int. 2005;68(2):515–28.
Wang-Rosenke Y, Neumayer HH, Peters H. NO signaling through cGMP in renal tissue fibrosis and beyond: key pathway and novel therapeutic target. Curr Med Chem. 2008;15(14):1396–406.
Stasch JP, Schlossmann J, Hocher B. Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr Opin Pharmacol. 2015;21:95–104.
Kalk P, Godes M, Relle K, Rothkegel C, Hucke A, Stasch JP, et al. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br J Pharmacol. 2006;148(6):853–9.
Wang Y, Kramer S, Loof T, Martini S, Kron S, Kawachi H, et al. Stimulation of soluble guanylate cyclase slows progression in anti-thy1-induced chronic glomerulosclerosis. Kidney Int. 2005;68(1):47–61.
Geschka S, Kretschmer A, Sharkovska Y, Evgenov OV, Lawrenz B, Hucke A, et al. Soluble guanylate cyclase stimulation prevents fibrotic tissue remodeling and improves survival in salt-sensitive Dahl rats. PLoS One. 2011;6(7), e21853.
Sharkovska Y, Kalk P, Lawrenz B, Godes M, Hoffmann LS, Wellkisch K, et al. Nitric oxide-independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low-renin and high-renin models. J Hypertens. 2010;28(8):1666–75.
Zoccali C. The endothelium as a target in renal diseases. J Nephrol. 2007;20 Suppl 12:S39–44.
Stasch JP, Hobbs AJ. NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol. 2009;191:277–308.
Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–40.
Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh A, Langleben D, Fritsch A, Menezes F et al.. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. 2015.
Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–29.
Simonneau G, D’Armini AM, Ghofrani HA, Grimminger F, Hoeper MM, Jansa P, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2). Eur Respir J. 2015;45(5):1293–302.
Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014;146(5):1274–85.
Bonderman D, Ghio S, Felix SB, Ghofrani HA, Michelakis E, Mitrovic V, et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. 2013;128(5):502–11.
Pieske B, Butler J, Filippatos G, Lam C, Maggioni AP, Ponikowski P, et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur J Heart Fail. 2014;16(9):1026–38.
Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: The SOCRATES-REDUCED Randomized Trial. JAMA. 2015;314(21):2251–62.
Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, et al. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124(25):2882–91.
Shah AM, Claggett B, Sweitzer NK, Shah SJ, Anand IS, Liu L, et al. Prognostic importance of impaired systolic function in heart failure with preserved ejection fraction and the impact of spironolactone. Circulation. 2015;132(5):402–14.
Kramann R, Erpenbeck J, Schneider RK, Rohl AB, Hein M, Brandenburg VM, et al. Speckle tracking echocardiography detects uremic cardiomyopathy early and predicts cardiovascular mortality in ESRD. J Am Soc Nephrol. 2014;25(10):2351–65.
Liu YW, Su CT, Sung JM, Wang SP, Su YR, Yang CS, et al. Association of left ventricular longitudinal strain with mortality among stable hemodialysis patients with preserved left ventricular ejection fraction. Clin J Am Soc Nephrol. 2013;8(9):1564–74.
Myerson SG, BelIenger NG, Pennell OJ. Assessment of left ventricular mass by cardiovascular magnetic resonance. Hypertension. 2002;39(3):750–5.
Acknowledgments
This work was supported by research grants from the National Institutes of Health (K23 DK092354 and R03 DK104013 [to RFD]; and R01 HL107577 and R01 HL127028 [to SJS]).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ruth F. Dubin declares that they have no conflict of interest.
Sanjiv J. Shah has received consulting fees from AstraZeneca, Bayer, Merck, and Novartis.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pharmacologic Therapy
Rights and permissions
About this article

Cite this article
Dubin, R.F., Shah, S.J. Soluble Guanylate Cyclase Stimulators: a Novel Treatment Option for Heart Failure Associated with Cardiorenal Syndromes?. Curr Heart Fail Rep 13, 132–139 (2016). https://doi.org/10.1007/s11897-016-0290-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-016-0290-z