
Abstract
Purpose of review
We provide an overview of pancreas pathology in type 1 diabetes (T1D) in the context of its clinical stages.
Recent findings
Recent studies of pancreata from organ donors with T1D and non-diabetic donors expressing T1D-associated autoantibodies reveal pathological changes/disease mechanisms beyond the well-known loss of β cells and lymphocytic infiltrates of the islets (insulitis), including β-cell stress, dysfunction, and viral infections. Pancreas pathology evolves through disease stages, is asynchronous, and demonstrates a chronic disease that remains active years after diagnosis. Critically, β-cell loss is not complete at onset, although young age is associated with increased severity.
Summary
The recognition of multiple pathogenic alterations and the chronic nature of disease mechanisms during and after the development of T1D inform improved clinical trial design and reveal additional targets for therapeutic manipulation, in the context of an expanded time window for intervention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease leading to severe loss of pancreatic β cells. The disease often manifests in children and adolescents, but many patients are diagnosed as adults [1, 2]. The prominent pancreas pathological features of T1D are loss of β cells and islet inflammation. The discovery of autoantibodies led to the recognition that autoimmunity may be triggered even in early life, and autoantibody conversion precedes clinical symptoms from months to years. All of the above and early pathology studies led to the belief that β-cell destruction is occurring over time, largely prior to the clinical onset, and that about 90% of the β cells are lost by the time symptoms manifest. Autoreactive T cells are considered the primary mediators of β-cell loss [3]. Since the mid-1980s, the design of clinical trials for preventing or reversing diabetes has been based on these views.
Here, we provide an updated view of pancreas pathology in T1D. We revisit earlier and recent studies to describe how our knowledge has evolved. Systematic efforts to provide greater access to the T1D pancreas to the scientific community, improved molecular methods, and collaboration have advanced our understanding of T1D pathogenesis and pathology, including the discovery of additional disease mechanisms, cellular players, and pathological features, all of which may be amenable to therapeutic manipulation. We also discuss current gaps in knowledge, which are especially critical during the prodromic phases of the disease, for which the characterization of pancreas pathology remains limited.
Sources of Human Pancreas for T1D Research
Access to the pancreas from patients with T1D has been historically limited, but it has been possible to obtain pancreata from patients through autopsy, biopsy, and organ donation. Currently, three pancreatic biobanks are actively supporting T1D research: the Exeter Archival Diabetes Biobank (EADB) in the UK, the Diabetes Virus Detection study (DiViD) in Norway, and the Network for Pancreatic Organ Donors with Diabetes (nPOD) in the USA. These are described below:
EADB
Studies of autopsy pancreas were first, reflecting the higher probability of patients passing away following complications of ketoacidosis, which are now rare with improved therapies [4,5,6]. Established by Foulis in the 1980s, the EADB holds formalin-fixed, paraffin-embedded pancreas blocks from nearly 200 patients, of which about half are from young patients (< 20 years old) with recent-onset T1D. Thus, the EADB is the world’s largest collection of autopsy pancreas samples recovered near a diagnosis of T1D.
DiViD
Percutaneous biopsies were performed in Japan in the 1990s [7, 8]; although safe overall, the approach yielded little material, which limits investigations and their significance given that only a small area of pancreas can be examined. In 2014, the DiViD study reported obtaining specimens via laparoscopic pancreatic tail resection from six living adult patients with newly diagnosed T1D (24–35 years old) [9]. A significant amount of tissue was obtained, and samples were shared with many investigators around the world for collaborative studies. However, surgical complications led to the closure of the study and no additional biopsies were performed [10].
nPOD
Established in 2007, nPOD has and continues to obtain pancreas and other tissues from organ donors with T1D and these are provided to the scientific community [11]. The T1D donors recovered cover a wide range of age and T1D duration. nPOD collects tissues from organ donors without diabetes and screens them to identify those with autoantibodies who might have been developing T1D. Thus, nPOD is attempting to obtain tissues that could inform about the preclinical stage of T1D. Samples available include tissues that are fixed, frozen, or fresh and are derived from the pancreas, spleen, pancreatic and non-pancreatic lymph nodes, blood (whole blood, serum, and plasma), duodenum, and thymus. Presently, nPOD is the largest biobank dedicated to T1D research; it has collected 185 non-diabetic donors, 36 autoantibody-positive (AAb+) donors, 168 donors with T1D, and donors with other forms of diabetes (T2D, MODY, GDM, cystic fibrosis).
Overall, these efforts have recovered pancreas from patients with T1D during the last 80 years, with varying age of onset and disease duration (Fig. 1a); these biobanks are complementary to each other and tremendously valuable for the T1D research community.

Key features of the patients in the EADB, DiViD, and nPOD biobanks. a Dot plot illustrating the differences in age at onset and disease duration for the different three main pancreas biobanks, EADB (black circles), DiViD (white triangles), and nPOD (white squares). The EADB cohort is enriched for young-onset, short-duration T1D cases, whereas the nPOD cohort contains many donors with older onset and longer disease duration. b Age of onset strongly determines the proportion of cases with residual ICIs > 1 year post-diagnosis. Bar graph shows the % of cases with > 1 year duration of disease with residual ICIs divided based on the age at diagnosis: < 7 years (13.5%, black bars), 7–12 years (26.7%, hatched bars), and > 13 years (49.2%, white bars). Sources: http://foulis.vub.ac.be/; https://www.jdrfnpod.org/for-investigators/online-pathology-information/; Krogvold et al. 2014 [12•]
Key Features of Pancreas Pathology in T1D
Studies by LeCompte [13], Gepts [4], and others provided initial insight onto pancreas pathology in T1D. When Gepts described the T1D pancreas pathology in 1965 [4], T1D was referred to as “juvenile diabetes” and classified as acute (near diagnosis) and chronic (long duration); the role of autoimmunity was unknown, and islet cell antibodies were not discovered until 1974 [14]. Gepts evaluated the pancreas from 40 patients, 22 of whom were considered to have acute juvenile diabetes as they passed soon after diagnosis (disease duration range 3–180 days, < 90 days for 21/22 patients); this was a cohort of young children (mean age 10.89 years; ten children below age 10, nine teens aged 13–17, and only three adults aged 21, 22, and 30). Gepts made the following observations in these young patients with recent onset diabetes: 1) a drastic reduction in the number of β cells, estimated at less than 10% of well, age-matched individuals without diabetes; 2) residual β cells with cytological signs of marked activity, presence of large islets, and signs of new islet formation; and 3) peri- and intra-insular inflammatory (termed “inflaminatory”) infiltrates in 68% of the patients. Gepts also evaluated pancreata from 18 patients with chronic diabetes (disease duration 2–37 years, mean 17 years) who were on average 12.5 years old at diagnosis. In this cohort, the inflammatory process was not observed but β cells were completely absent, with few exceptions. Thus, islet inflammation and β-cell loss have been considered the main pathological features of T1D. The other major pathological feature reported in the T1D pancreas is the expression of elevated levels of HLA class I molecules, both in the cytoplasm and on the surface of islet cells, first reported by Bottazzo and Foulis in the mid-1980s [15,16,17]. These are considered the most typical features of T1D pancreas pathology and are reviewed below.
Insulitis
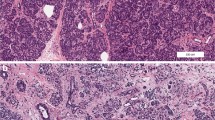
This pathognomonic lesion consists of immune cell infiltrates within and around the pancreatic islets [18], and it supports the concept that T1D is a T-cell-mediated autoimmune disease. In the mid-1980s, expanding on the studies by Gepts, Foulis et al. [6] reported insulitis in 78% of young patients with recent onset disease (< 1 year). A 2011 meta-analysis by In’t Veld [18] collected information from studies published since 1902 (213 cases with insulitis) and reported that insulitis occurs in 73% of young (< 14 years) patients with T1D who have a short duration of disease (< 1 month), in 60% of young patients with disease duration between 1 month and 1 year, and only in 4% of young patients with a duration of disease longer than 1 year. This scenario drastically changes in older patients. Only 29% of cases with onset between 15 and 40 years of age and disease duration < 1 month showed insulitis. Foulis reported that 23% of insulin-containing islets (ICIs) and only 1% of insulin-deficient islets (IDIs) had insulitis in young patients with < 1 year disease duration. Willcox and colleagues [19] examined the pancreas of 29 young patients (mean age 11.7 years) with disease duration between 1 day and 18 months; 23.8% of the islets contained insulin, of which 34.8% had insulitis, including 5% of IDIs. In the meta-analysis by In’t Veld involving young patients with recent-onset disease (< 1 month), 34% of the islets stained for insulin on average, but only 33.6% of these islets had insulitis; in older patients with recent-onset disease, an average of 63% of the islets contained β cells, with 18% of the insulin-containing islets also showing insulitis [18]. In the DiViD study, the proportion of islets with insulitis ranged between 5 and 58% and only a single patient had insulitis in more than 50% of the islets [9, 12•]; in these adult patients, an average of 11% of the islets examined showed insulitis. In a study of nPOD organ donors with variable duration of T1D, 17 had insulitis with a broad range of disease duration (0–12 years) and age of onset (4–28 years); importantly, the frequency of insulitis had limited inverse correlation with diabetes duration and no correlation with age, whether at diagnosis or passing. Thus, the proportion of islets showing insulitis in the T1D pancreas is, overall, moderate to low; however, it varies significantly with age and disease duration. It is evident from the above that insulitis can be observed in many patients many years after diagnosis [20•].
According to the 2013 consensus [21], insulitis is defined by a predominantly lymphocytic infiltration of the islets consisting of at least 15 CD45+ cells/islet (Fig. 2a) in a minimum of three islets, and the pancreas should also contain presence of IDIs or pseudoatrophic islets. Inflammatory infiltrates are more commonly detected in the islet periphery (peri-insulitis) or within the islet, with peri-insulitis representing the predominant form. Insulitis in the human pancreas is therefore much less severe than in experimental mouse models, in which a large number of infiltrating cells can be found in the majority of the islets. Insulitis is typically found in ICIs and less commonly in pseudoatrophic islets. Both T and B lymphocytes are present. Cytotoxic CD8+ T cells represent the predominant lymphocyte populations; nPOD studies demonstrated that at least a proportion of the CD8+ T cells are autoreactive and target β-cell autoantigens; the diversity in the antigen specificity of the infiltrating CD8+ T cells was higher in patients with longer disease duration, suggesting that autoimmunity evolves even after diagnosis [22••]. Other cell types commonly detected in the insulitis lesion are B lymphocytes, macrophages, and CD4+ T cells [19]. The analysis of 21 patients (1 day–6 months’ duration, median age 12 years) demonstrated two distinct patterns of infiltration: one characterized by large numbers of infiltrating cells, especially CD20+ B lymphocytes, defined as CD20 high (CD20hi; Fig. 2b); the second pattern was characterized by infiltrates with fewer cells, including less CD20+, defined as CD20 low (CD20lo; Fig. 2c) [23•]. CD20hi subjects had a lower number of ICIs and they were younger (mean of 7.8 years) when compared to CD20lo subjects (mean of 13 years). The association of insulitis lesions containing higher proportions of B lymphocytes with younger age at diagnosis suggests that these cells may contribute to a more aggressive form of autoimmunity [23•, 24]. Of importance is also the fact that all of the inflamed islets within a given patient display the same insulitic profile but that this profile differed significantly between individuals [23•].

Key features of islets from type 1 diabetes donors. a A representative T1D donor islet with insulitis (DiViD3); insulin (light blue), CD45 (green), and DAPI (dark blue). Representative islet from a CD20Lo case (DiViD2) (b) and a CD20Hi donor (nPOD 6209) (c); insulin (light blue), CD20 (green), CD8 (red), and DAPI (dark blue). Images courtesy of P. Leete (University of Exeter). d Expression of HLA class I and Enteroviral VP1 in an ICI from a T1D donor (EADB E560); insulin (light blue), VP1 (green, arrows), HLAI (red), and DAPI (dark blue)
β-Cell Destruction
The most striking pathological feature in the T1D pancreas is loss of β cells. Lack of insulin staining is the predominant feature, and it is severe in the pancreas from patients who had T1D for many years. There is also substantial loss by the time of diagnosis, yet the long-held belief that 90% of the β-cell mass is universally lost at diagnosis is no longer supported. Consistent with the findings of Gepts, studies from the EADB, nPOD, DiViD cohorts, and other collections [25, 26] support that younger children have more severe loss of β cells (Table 1); however, patients who develop T1D when teenagers or older may still have 40–60% of their islets containing β cells and staining positive for insulin at diagnosis [23•, 26]. Accordingly, the DiViD biopsies of six adult patients demonstrated insulin staining, on average, in 36% of the islets (range 18–66%) [12•]. Among 80 nPOD donors with T1D, of whom only a few had disease duration less than 1 year [20•], residual β cells were observed in all T1D donors with insulitis, who had a 10-fold higher β-cell mass compared to those without insulitis. By contrast, the analysis by Leete et al. [23•] of 20 young patients (mean age 10.5 years) who died within 3 months of diagnosis showed much more severe β-cell loss; moreover, this varied according to the insulitis pattern, CD20lo and CD20hi; the ratio CD20 to CD4 also varied consistently with the two phenotypes (> 1 in CD20hi and < 1 in CD20lo). This ratio led to the separation of all individuals into three different groups: 1) < 7 years (CD20hi), 2) 7–12 years, and 3) ≥ 13 years (CD20lo). Group 1 retained less ICIs than groups 2 and 3. Strikingly, the proportion of residual ICIs in those diagnosed early in life was around 14% while those diagnosed from their teens or beyond had higher number of insulin-positive islets (39% ICIs). Combination of all the available data for the EADB and nPOD cases with > 1 year duration of disease (Fig. 1b) demonstrates that ICIs are preferentially retained in the older onset, group 3 cases. Thus, the emerging evidence suggests that β-cell destruction is quite heterogeneous but greater loss is associated with younger onset of disease, the autoimmune process affects only a moderate proportion of islets at any given time, and it continues for several years after diagnosis.
Hyperexpression of HLA-Class I Molecules by Islet Cells
The elevated levels of HLA class I molecules (Figs. 2d and 3) in islet cells highlight an inflammatory state and it is often associated with insulitis [33]. Like insulitis, hyperexpression of HLA class I molecules is typically found in ICIs, and it is often associated with CD8+ T-cell infiltrates. It is possible that β cells hyperexpressing HLA class I molecules present their self-antigens to autoreactive T cells. Hyperexpression of class I molecules may result from viral infections associated with T1D [30•, 34], but it is unknown whether infiltrating CD8+ T cells target viral epitopes presented by infected β cells [35]. Like insulitis, this phenomenon continues to be present for several years after diagnosis, and it has been validated with multiple approaches at the protein and RNA levels [28•]. A 2018 study classified patients based on a urinary C-peptide/creatinine ratio regression model and revealed that C-peptide loss continues for the first 7 years post-diagnosis but C-peptide levels stabilize afterwards, suggesting that from then on residual β cells are no longer being actively destroyed [27•]. To ascertain if the decline in hyperexpression of HLA class I correlates with this phenomenon, we combined data from the EADB [15] and Richardson et al. [28•] and found that HLA class I hyperexpression was not restricted only to recent-onset patients but also in those with longer disease duration who had residual ICIs. However, the proportion of residual ICIs hyperexpressing HLA class I clearly decreased over time. In patients with T1D for < 7 years, almost all ICIs hyperexpressed HLA class I molecules in contrast to only a median of 14% (0.0–52.3) among those who had T1D for > 7 years (Table 1). In summary, HLA class I hyperexpression persists on the majority of residual ICIs within the first 7 years post-diagnosis and this may contribute to β-cell demise by facilitating the presentation of self-peptides to infiltrating autoreactive CD8+ T cells. As this hyperexpression declines in long-standing disease, β-cell antigen presentation would be attenuated, potentially leading to a reduction in the rate of destruction, even in the face of low-level, persistent insulitis.

Immunofluorescence analysis of HLA-I expression in frozen pancreas from a patient with type 1 diabetes (disease duration, 7 years) from the nPOD cohort. a Hyperexpression of HLA-I (red) can be predominantly seen in ICIs islets (green). Scale in whole tissue image = 2000 μm. b, c Higher magnification of the inset from (a), with (b) or without DAPI counterstain (c); scale in zoomed image = 200 μm
Novel Pathology Findings in the T1D Pancreas
Studies are demonstrating additional pancreatic pathological abnormalities. Among these are changes in extracellular matrix components. Accumulation of hyaluronan (HA), a key constituent of the extracellular matrix, and HA binding proteins is found around islet cells and infiltrating lymphocytes in islets affected by insulitis [36]. HA deposits occur along the edge capillaries of diabetic islets, where leukocyte infiltrates in insulitis are frequently observed, and along intra-islet microvessels. HA deposition is more pronounced in islets from younger donors with T1D and those examined within the first year from diagnosis, confirming a more aggressive pathology in these patients. Conversely, the morphological pattern of HA in insulitis-free pancreas from donors with long-standing diabetes is similar to normal islets [36]. These studies indicate that HA and proteins associated with it form a matrix that interacts with infiltrating cells and it is directly related to pancreatic β-cell loss and insulitis [32]. HA might create a permissive environment that favors autoimmunity by restricting regulatory T-cell differentiation [37, 38], thus favoring effector T cells. Treatment of NOD mice with an inhibitor of HA synthesis, 4-methylumbelliferone (4-MU), inhibited progression to diabetes and increased the ratio of regulatory T cells to T effector cells [38]. Immunohistochemistry for laminin, perlecan, and collagen shows that components of the peri-islet basal membrane are lost at sites of leukocyte infiltration of the islets [39]. This indicates that removal of the basal membrane takes place during leukocyte entry into the islets. Moreover, cathepsins were found in the insulitis lesion near areas of disruption of the peri-islet basement membrane and may favor the penetration of lymphocytes inside the islets [39]. Alterations of extracellular matrix components also impact β-cell function and survival [40], including the loss of heparan sulfate which is associated with β-cell apoptosis [41].
Emerging studies are providing growing support for a viral contribution to the disease pathogenesis, particularly by enteroviruses [42]. Enterovirus proteins, enterovirus RNA, and an active anti-viral host response have been demonstrated in the pancreata of T1D donors from each of the three biobanks [43]. Viral capsid protein is detected in a small number of β cells, typically only in ICIs, often in association with hyperexpression of HLA class I molecules and insulitis (Fig. 2d) [34, 44]. Enterovirus infections can severely impair insulin secretion [45], impact gene expression and microRNA regulation [46], and induce inflammation.
In turn, inflammation promotes β-cell stress, protein misfolding, dysfunction, and apoptosis [31, 47,48,49]. Indeed, there is growing evidence, also at the pathology level, that residual β cells in the T1D pancreas exhibit multiple signs of cellular stress, including an increased expression of ER stress markers [29, 50], especially in infiltrated ICIs [29, 51], which may contribute to insulin secretion abnormalities during the prediabetic phase [52]. β-cell dysfunction may be an important contributor to insulin deficiency also at onset, when, as discussed above, many patients would be likely to have a significant residual β-cell mass [29, 53,54,55,56]. Moreover, islet function may be recoverable [53]; in the DiViD study, islets isolated from pancreas biopsies from newly diagnosed patients recovered function in culture. There is also evidence for dysregulated sphingolipid metabolism [57] and altered proteomic profiles that involve inflammatory, immune, and metabolic pathways [58]. β-cell inflammation and stress may favor the formation of post-translationally modified and hybrid autoantigen peptides which may have a critical role in breaking self-tolerance and triggering islet autoimmunity [59]. For example, endoplasmic reticulum stress alters the endomembrane distribution of GAD65 autoantigen, resulting in accumulation of a more immunogenic, palmitoylated form of this molecule in trans-Golgi membranes, as demonstrated by the pathological examination of nPOD donors [60].
The exocrine pancreas is also impacted in T1D: the pancreas of donors with T1D is only 55% the weight of that of donors without diabetes [61]; this reduction in weight primarily affects the pancreatic dorsal lobe, which includes the majority of the head and the entire body and tail. Such a reduction is observed close to onset, and T1D donors with long disease duration have almost normal pancreatic weights. There is initial evidence that non-diabetic, autoantibody-positive nPOD donors with insulitis have a small decrease in pancreas weight [62]. Pancreas volume, volume normalized by body weight, volume normalized by body mass index, and volume normalized by body surface area were all lower in patients with T1D compared to controls according to imaging studies [63]. As the islets constitute only 1–2% of the pancreas volume, these findings suggest loss of exocrine tissue during the development of T1D. This is consistent with impaired exocrine function, which is reported at T1D diagnosis (low levels of elastase in stools) but not at the time of seroconversion to islet autoantibody positivity [64]. Morphometric studies show that T1D donors have a higher non-exocrine–non-endocrine tissue area to total pancreas area than non-diabetic controls regardless of age, suggesting that T1D affects the entire pancreas [65]. In addition, large numbers of infiltrating cells have been found in the exocrine pancreas; CD8+ and CD4+ T cells, and CD11c+ cells, were present in high numbers in the exocrine pancreas of AAb+ and recent-onset T1D donors, with a predominance of CD8+ T cells and no reported differences between donors with or without pancreatitis [66]. The phenotype and function of these cells remains unclear. Mohapatra et al. created the term “diabetic exocrine pancreatopathy” to define the moderate-to-severe subclinical pancreatic fibrosis and modest exocrine dysfunction in the absence of clinical or histopathological evidence of chronic pancreatitis that affects individuals with T1D [67]. This includes (1) markedly decreased pancreatic weight, size, and volume; (2) increased inter-acinar fibrosis and acinar atrophy with minimal inflammation and no pancreatic ductal changes; (3) reduced exocrine enzyme output and fecal elastase concentrations; (4) normal to minimal decrease in coefficient of fat absorption; and (5) lack of progression of exocrine dysfunction over time.
Pancreas Pathology During Preclinical Disease Stages
Progress has been made toward understanding the natural history of islet autoimmunity from the longitudinal evaluation of relatives or individuals carrying HLA alleles associated with increased T1D risk. The best predictor of future T1D is the detection of circulating autoantibodies to islet autoantigens. Autoantibodies are found in almost 95% of those who develop clinical symptoms of T1D [68]. Longitudinal studies of large birth cohorts at increased genetic risk of T1D have shown that there is a peak in islet autoimmunity at 2–5 years of age; in these young children, progression to clinical disease is faster than those who convert at older age [69,70,71]. The highest risk is observed in those with autoantibodies against multiple islet autoantigens, in whom risk of T1D is about 40% at 5 years, 70% at 10 years, and 85% at 15 years [70]; however, those with a single autoantibody have much lower risk, around 5–10%, even with long follow-up. Studies have shown that metabolic abnormalities and defects in insulin secretion (assessed by C-peptide levels during an oral glucose tolerance test) become evident late in the progression to clinical disease, typically 18 to 6 months before diagnosis [72]. Based on the above, the JDRF, the Endocrine Society, and the American Diabetes Association have recognized three different stages in the progression of islet autoimmunity toward clinical T1D [73], which are 1) stage 1, defined by the presence of two or more autoantibodies; 2) stage 2, in which glucose intolerance or dysglycemia are also present; and 3) stage 3, which represents clinically manifest diabetes, when classical symptoms (polyuria, polydipsia, fatigue, and diabetic ketoacidosis) and laboratory evidence of severe, fasting hyperglycemia are present. However, an earlier stage not formally recognized by this classification is characterized by the presence of a single autoantibody. We have discussed the pathology of stage 3 in the preceding sections; here, we will review what is known about pancreas pathology in the preclinical stages of T1D.
Pathology Findings at Stages 1 and 2 in Single Versus Multiple AAb Positivity
There is little information as to whether pathological alterations are different at stages 1 and 2 of the clinical classification because too few donors have been studied so far. Despite the autoantibody screening of organ donors instituted by nPOD, the number of autoantibody-positive donors, especially those with multiple autoantibodies, and more so those who also have elevated HbA1c levels, is quite low in the general population. Moreover, only a fraction of the autoantibody-positive donors is recovered, as many of these pancreata are allocated to transplantation instead of research; we advocate that these rare donors should be allocated to research [74••]. Furthermore, not all AAb+ donors, especially those with a single autoantibody, may represent true prediabetic individuals who would have developed T1D. Functional assessment of donor pancreas is just beginning through the study of isolated islets from donors with T1D [75, 76], pioneered by nPOD, which will be applied in the future to the pancreas from autoantibody-positive donors. Functional assessment of islet function in pancreas slices [77], which allows examining islet function in the natural tissue environment, is predicted to reveal novel information in the next few years. Sustained efforts may allow the identification of pathological features that define stage 1 and stage 2 T1D.
However, it is possible to examine pancreas pathology and contrast findings in donors with single versus multiple autoantibodies, with single autoantibody positivity representing the earlier phase in the natural history of islet autoimmunity and those with multiple autoantibodies representing donors at stage 1, or 2, if they had elevated HbA1c. Gianani et al. [78] identified a donor with a single autoantibody with no reduction in β-cell area and no insulitis. One of the largest studies identified AAb+ donors [79] by screening donors whose pancreata were used for islet isolation. A total of 1507 donors (25–60 years old) were identified; 55 of these had a single autoantibody, 4 donors had two, 2 donors had three, and 1 donor had four autoantibodies. Of these, only two of the triple autoantibody-positive donors had insulitis and carried high-risk HLA types; a small percentage of islets (9 and 3%, respectively) had insulitis. In both, at least one insulin-negative islet could be found. However, there was no decrease in β-cell mass. Another screening identified 32 autoantibody-positive donors among 969 tested (3.3%): nine expressed multiple autoantibodies but none carried high-risk HLA types and insulitis was not observed [80]. In both studies, the amount of tissue available was limited to a small tissue block, and thus sampling issues cannot be excluded.
So far, nPOD [20•] has reported 21 donors with a single autoantibody, usually in the absence of T1D-associated HLA genes in whom insulitis was absent. However, 2/6 donors with multiple autoantibodies and 1 donor with a single autoantibody had insulitis and T1D-associated HLA types. Another study of nPOD donors reported the CD8+ T cells tended to be higher in both islet and exocrine areas in some AAb+ donors than controls; the AAb+ group was the only one in addition to T1D donors with remaining ICIs in which the ratio between endocrine and exocrine infiltration was elevated, suggesting a polarization of CD8+ T cells toward the islets [66]. In these two studies, there were no statistically significant differences in β-cell area or mass between non-diabetic and AAb+ individuals. However, AAb+ individuals with insulitis showed higher β-cell area than their non-insulitic counterparts and a slight increase in islet area compared to non-diabetic donors [81] was also reported. Perhaps these findings are consistent with the enlarged islets and features of hyperactivity originally reported by Gepts [4].
As noted, hyperexpression of HLA class I molecules in ICIs is a feature of T1D and has been observed in the EADB, nPOD, and DiViD cohorts. It was also observed in double AAb+ donors [82]; around 13% of the islets showed HLA class I hyperexpression in head, body, and tail of the pancreas with no particular distribution. Areas of islets with normal HLA class I expression were frequently contiguous to areas with hyperexpression. CD8+ T-cell infiltration, although mild, was on average higher in islets with high HLA class I compared to islets with normal expression, consistent with the hypothesis that HLA class I expression could attract cytotoxic T cells to the islets. Ongoing studies by the nPOD-Virus group are screening non-diabetic, single, double AAb+, and T1D donors for the expression of HLA class I molecules together with other markers of viral infection in an attempt to study a possible association with enterovirus infections.
Despite the low number of AAb+ donors analyzed, these studies demonstrate islet pathological changes at stage 1 since insulitis can be found in less than half of the donors with multiple autoantibodies; however, so far it appears that only a limited proportion of islets may show concomitant β-cell loss. Insulitis does not appear in donors with a single autoantibody. While the number of subjects examined cannot be considered sufficient to draw firm conclusions, it appears that the single autoantibody stage may not be associated with the key features of the T1D pancreas pathology.
Stage 2: Multiple AAb and Impaired Glucose Tolerance
As noted, at present there is no published study that specifically examines pancreas pathology in individuals with multiple autoantibodies and elevated HbA1c. We speculate that at this stage insulitis and β-cell loss may become more prominent; moreover, the increase in insulin demand may exceed the ability of β cells to process newly translated proteins, leading to the accumulation of unfolded proteins [52, 83]. As discussed above, this promotes ER stress and apoptosis [29, 51], which precedes clinical onset. A key sign of β-cell ER dysfunction is the accumulation of unprocessed proinsulin [51], which is released to the circulation. This produces an increase in the proinsulin to C-peptide ratio in the serum of at-risk individuals months prior to diagnosis [52, 84]. At the pancreas pathology level, there is an increase in proinsulin and in the proinsulin/insulin ratio in the pancreas of double AAb+ nPOD donors and, importantly, in some with a single autoantibody [81].
Conclusions
The major features of T1D pancreas pathology highlight the chronicity of the disease, as its key pathological features are demonstrated for several years after diagnosis, and to some extent before diagnosis. Critically, insulitis, β-cell loss, and hyperexpression of HLA class I molecules do not affect all islets at the same time. Metabolic testing of living patients at diagnosis demonstrates severe but not complete impairment of stimulated C-peptide responses [85,86,87,88,89], with further decline in the following years; typically, decline is more severe in younger children. When examining the pancreas, the severity of β-cell loss at diagnosis is variable but not as high as previously believed, and several studies have demonstrated low amount of C-peptide and a response to stimulation in patients who had T1D for decades [90,91,92,93,94]. The persistence of β cells even decades after diagnosis with evidence of low-level replication [91], and the growing evidence for inflammation and ER stress [50, 95] imply that β-cell dysfunction plays a significant role in causing the symptoms at the time of diagnosis and probably for a few years thereafter. Recent pathology studies have shown that β-cell destruction is often incomplete at onset and continues after diagnosis for several years; besides T-cell-mediated autoimmunity, there are additional pathological alterations for which therapeutic manipulation is possible. Overall, the therapeutic time window for intervention may be longer than previously thought, and intervention strategies should have broader scope to simultaneously target multiple disease pathways that pathology studies show to be asynchronously active at any given time.
Abbreviations
- AAb+:
-
Autoantibody positive
- EADB:
-
Exeter Archival Diabetes Biobank
- ER:
-
Endoplasmic reticulum
- DiViD:
-
Diabetes Virus Detection Study
- GAD:
-
Glutamic acid decarboxylase
- HA:
-
Hyaluronan
- HLAI:
-
Human leukocyte antigen class I
- IA-2:
-
Islet antigen-2
- ICI:
-
Insulin-containing islet
- IDI:
-
Insulin-deficient islet
- MODY:
-
Maturity onset diabetes of the young
- NOD:
-
Non-obese diabetic mouse
- nPOD:
-
Network for Pancreatic Organ Donors with Diabetes
- T2D:
-
Type 2 diabetes
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross-sectional, genetically stratified survival analysis from UK biobank. Lancet Diabetes Endocrinol. 2018;6(2):122–9.
Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinol Metab Clin N Am. 2010;39(3):481–97.
Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. 2017;127(8):2881–91.
Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–33.
Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes. 1978;27(Supplement 1):251–61.
Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29(5):267–74.
Hanafusa T, Miyazaki A, Miyagawa J, Tamura S, Inada M, Yamada K, et al. Examination of islets in the pancreas biopsy specimens from newly diagnosed type 1 (insulin-dependent) diabetic patients. Diabetologia. 1990;33(2):105–11.
Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92(5):2313–22.
Krogvold L, Edwin B, Buanes T, Ludvigsson J, Korsgren O, Hyoty H, et al. Pancreatic biopsy by minimal tail resection in live adult patients at the onset of type 1 diabetes: experiences from the DiViD study. Diabetologia. 2014;57(4):841–3.
Atkinson MA. Pancreatic biopsies in type 1 diabetes: revisiting the myth of Pandora’s box. Diabetologia. 2014;57(4):656–9.
Pugliese A, Yang M, Kusmarteva I, Heiple T, Vendrame F, Wasserfall C, et al. The Juvenile Diabetes Research Foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) Program: goals, operational model and emerging findings. Pediatr Diabetes. 2014;15(1):1–9.
• Krogvold L, Wiberg A, Edwin B, Buanes T, Jahnsen FL, Hanssen KF, et al. Insulitis and characterisation of infiltrating T cells in surgical pancreatic tail resections from patients at onset of type 1 diabetes. Diabetologia. 2016;59(3):492–501. This article reports obtaining pancreas tail biopsies from living patients with new onset T1D in the DiViD study.
Lecompte PM. Insulitis in early juvenile diabetes. AMA Arch Pathol. 1958;66(4):450–7.
Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974;2(7892):1279–83.
Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1987;30(5):333–43.
Pujol-Borrell R, Todd I, Londei M, Foulis A, Feldmann M, Bottazzo GF. Inappropriate major histocompatibility complex class II expression by thyroid follicular cells in thyroid autoimmune disease and by pancreatic beta cells in type I diabetes. Mol Biol Med. 1986;3(2):159–65.
Bottazzo GF, Dean BM, McNally JM, Mackay EH, Swift PGF, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. New Engl J Med. 1985;313:353–60.
In't Veld P. Insulitis in human type 1 diabetes: the quest for an elusive lesion. Islets. 2011;3(4):131–8.
Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. ClinexpImmunol. 2009;155(2):173–81.
• Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and beta-cell mass in the natural history of type 1 diabetes. Diabetes. 2016;65(3):719–31. The study reports the characterization of insulitis and β-cell mass in nPOD donors across a spectrum of ages and disease duration.
Campbell-Thompson ML, Atkinson MA, Butler AE, Chapman NM, Frisk G, Gianani R, et al. The diagnosis of insulitis in human type 1 diabetes. Diabetologia. 2013;56(11):2541–3.
•• Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209(1):51–60. This study of nPOD donors demonstrates that islet-infiltrating CD8 + T cells are autoreactive.
• Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, et al. Differential insulitic profiles determine the extent of beta-cell destruction and the age at onset of type 1 diabetes. Diabetes. 2016;65(5):1362–9. Study of the EADB cohort reporting different insulitis profiles according to the abundance of CD20 + B lymphocytes.
Morgan NG. Bringing the human pancreas into focus: new paradigms for the understanding of type 1 diabetes. Diabet Med. 2017;34(7):879–86.
Klinke DJ. Age-corrected beta cell mass following onset of type 1 diabetes mellitus correlates with plasma C-peptide in humans. PLoS One. 2011;6(11):e26873.
Klinke DJ. Extent of beta cell destruction is important but insufficient to predict the onset of type 1 diabetes mellitus. PLoS One. 2008;3(1):e1374.
• Shields BM, McDonald TJ, Oram R, Hill A, Hudson M, Leete P, et al. C-peptide decline in type 1 diabetes has two phases: an initial exponential fall and a subsequent stable phase. Diabetes Care, 2018. 41(7):1486–92. This study reports that loss of C-peptide plateaus 7 years after diagnosis, with implications for future interventions and correlations with pathology findings.
• Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. 2016;59(11):2448–58. Joint study of the EADB, nPOD, and DiViD cohort defines hyperexpression of HLA class I molecules as a defining feature of T1D pathology using a multitude of methodologies.
Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55(9):2417–20.
• Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, et al. Detection of a low-grade enteroviral infection in the islets of Langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes. 2015;64(5):1682–7. DiViD study reporting evidence for low-grade enterovirus infections in the pancreas from patients with recent onset T1D.
de Beeck AO, Eizirik DL. Viral infections in type 1 diabetes mellitus—why the beta cells? Nat Rev Endocrinol. 2016;12(5):263–73.
Bogdani M. Thinking outside the cell: a key role for hyaluronan in the pathogenesis of human type 1 diabetes. Diabetes. 2016;65(8):2105–14.
Morgan NG, Leete P, Foulis AK, Richardson SJ. Islet inflammation in human type 1 diabetes mellitus. IUBMB Life. 2014;66(11):723–34.
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52(6):1143–51.
Kundu R, Knight R, Dunga M, Peakman M. In silico and ex vivo approaches indicate immune pressure on capsid and non-capsid regions of coxsackie B viruses in the human system. PLoS One. 2018;13(6):e0199323.
Bogdani M, Johnson PY, Potter-Perigo S, Nagy N, Day AJ, Bollyky PL, et al. Hyaluronan and hyaluronan binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis. Diabetes. 2014;27
Kuipers HF, Rieck M, Gurevich I, Nagy N, Butte MJ, Negrin RS, et al. Hyaluronan synthesis is necessary for autoreactive T-cell trafficking, activation, and Th1 polarization. Proc Natl Acad Sci U S A. 2016;113(5):1339–44.
Nagy N, Kaber G, Johnson PY, Gebe JA, Preisinger A, Falk BA, et al. Inhibition of hyaluronan synthesis restores immune tolerance during autoimmune insulitis. J Clin Invest. 2015;125(10):3928–40.
Korpos E, Kadri N, Kappelhoff R, Wegner J, Overall CM, Weber E, et al. The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes. 2013;62(2):531–42.
Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L, Wight TN. Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr Diab Rep. 2014;14(12):552.
Simeonovic CJ, Popp SK, Starrs LM, Brown DJ, Ziolkowski AF, Ludwig B, et al. Loss of intra-islet heparan sulfate is a highly sensitive marker of type 1 diabetes progression in humans. PLoS One. 2018;13(2):e0191360.
Richardson SJ, Morgan NG. Enteroviral infections in the pathogenesis of type 1 diabetes: new insights for therapeutic intervention. Curr Opin Pharmacol. 2018;43:11–9.
Morgan NG, Richardson SJ. Enteroviruses as causative agents in type 1 diabetes: loose ends or lost cause? Trends Endocrinol Metab. 2014;25(12):611–9.
Richardson SJ, Leete P, Bone AJ, Foulis AK, Morgan NG. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia. 2013;56(1):185–93.
Gallagher GR, Brehm MA, Finberg RW, Barton BA, Shultz LD, Greiner DL, et al. Viral infection of engrafted human islets leads to diabetes. Diabetes. 2015;64(4):1358–69.
Kim KW, Ho A, Alshabee-Akil A, Hardikar AA, Kay TW, Rawlinson WD, et al. Coxsackievirus B5 infection induces dysregulation of microRNAs predicted to target known type 1 diabetes risk genes in human pancreatic islets. Diabetes. 2016;65(4):996–1003.
Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25–53.
Brozzi F, Eizirik DL. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci. 2016;121(2):133–9.
Marroqui L, Lopes M, dos Santos RS, Grieco FA, Roivainen M, Richardson SJ, et al. Differential cell autonomous responses determine the outcome of coxsackievirus infections in murine pancreatic alpha and beta cells. elife. 2015;4:e06990.
Eizirik DL, Coomans de Brachene A. Checks and balances—the limits of beta-cell endurance to ER stress. Diabetes. 2017;66(6):1467–9.
Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013;56(2):234–41.
Sims EK, Chaudhry Z, Watkins R, Syed F, Blum J, Ouyang F, et al. Elevations in the fasting serum proinsulin-to-C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care. 2016;39(9):1519–26.
Krogvold L, Skog O, Sundstrom G, Edwin B, Buanes T, Hanssen KF, et al. Function of isolated pancreatic islets from patients at onset of type 1 diabetes: insulin secretion can be restored after some days in a nondiabetogenic environment in vitro: results from the DiViD study. Diabetes. 2015;64(7):2506–12.
Burch TC, Morris MA, Campbell-Thompson M, Pugliese A, Nadler JL, Nyalwidhe JO. Proteomic analysis of disease stratified human pancreas tissue indicates unique signature of type 1 diabetes. PLoS One. 2015;10(8):e0135663.
Grzesik WJ, Nadler JL, Machida Y, Nadler JL, Imai Y, Morris MA. Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J Clin Endocrinol Metab. 2015;100(3):E387–95.
Imai Y, Dobrian AD, Morris MA, Taylor-Fishwick DA, Nadler JL. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia. 2016;59(4):673–8.
Holm LJ, Krogvold L, Hasselby JP, Kaur S, Claessens LA, Russell MA, et al. Abnormal islet sphingolipid metabolism in type 1 diabetes. Diabetologia. 2018;61(7):1650–61.
Nyalwidhe JO, Grzesik WJ, Burch TC, Semeraro ML, Waseem T, Gerling IC, et al. Comparative quantitative proteomic analysis of disease stratified laser captured microdissected human islets identifies proteins and pathways potentially related to type 1 diabetes. PLoS One. 2017;12(9):e0183908.
Marre ML, James EA, Piganelli JD. beta cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol. 2015;3:67.
Phelps EA, Cianciaruso C, Michael IP, Pasquier M, Kanaani J, Nano R, et al. Aberrant accumulation of the diabetes autoantigen GAD65 in Golgi membranes in conditions of ER stress and autoimmunity. Diabetes. 2016;65(9):2686–99.
Campbell-Thompson ML, Kaddis JS, Wasserfall C, Haller MJ, Pugliese A, Schatz DA, et al. The influence of type 1 diabetes on pancreatic weight. Diabetologia. 2016;59(1):217–21.
Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, Kaddis JS. Pancreas organ weight in individuals with disease-associated autoantibodies at risk for type 1 diabetes. JAMA. 2012;308(22):2337–9.
Virostko J, Hilmes M, Eitel K, Moore DJ, Powers AC. Use of the electronic medical record to assess pancreas size in type 1 diabetes. PLoS One. 2016;11(7):e0158825.
Kondrashova A, Nurminen N, Lehtonen J, Hyoty M, Toppari J, Ilonen J, et al. Exocrine pancreas function decreases during the progression of the beta-cell damaging process in young prediabetic children. Pediatr Diabetes. 2018;19(3):398–402.
Bonnet-Serrano F, Diedisheim M, Mallone R, Larger E. Decreased alpha-cell mass and early structural alterations of the exocrine pancreas in patients with type 1 diabetes: an analysis based on the nPOD repository. PLoS One. 2018;13(1):e0191528.
Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes. 2014;63(11):3880–90.
Mohapatra S, Majumder S, Smyrk TC, Zhang L, Matveyenko A, Kudva YC, et al. Diabetes mellitus is associated with an exocrine pancreatopathy: conclusions from a review of literature. Pancreas. 2016;45(8):1104–10.
Wenzlau JM, Hutton JC. Novel diabetes autoantibodies and prediction of type 1 diabetes. Curr Diab Rep. 2013;13(5):608–15.
Achenbach P, Bonifacio E, Koczwara K, Ziegler AG. Natural history of type 1 diabetes. Diabetes. 2005;54(Suppl 2):S25–31.
Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013;309(23):2473–9.
Krischer JP, Lynch KF, Schatz DA, Ilonen J, Lernmark A, Hagopian WA, et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia. 2015;58(5):980–7.
Sosenko JM, Skyler JS, Beam CA, Krischer JP, Greenbaum CJ, Mahon J, et al. Acceleration of the loss of the first-phase insulin response during the progression to type 1 diabetes in diabetes prevention trial-type 1 participants. Diabetes. 2013;62(12):4179–83.
Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 2015;38(10):1964–74.
•• Burke GW 3rd, Posgai AL, Wasserfall CH, Atkinson MA, Pugliese A. Raising awareness: the need to promote allocation of pancreata from rare nondiabetic donors with pancreatic islet autoimmunity to type 1 diabetes research. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2016;17(1):306–7. This article advocates for allocating pancreata from non-diabetic donors with autoantibodies to research, to help obtain organs that inform about pancreas pathology in the preclinical disease stages.
Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, et al. Alpha cell function and gene expression are compromised in type 1 diabetes. Cell Rep. 2018;22(10):2667–76.
Chmelova H, Cohrs CM, Chouinard JA, Petzold C, Kuhn M, Chen C, et al. Distinct roles of beta-cell mass and function during type 1 diabetes onset and remission. Diabetes. 2015;64(6):2148–60.
Marciniak A, Cohrs CM, Tsata V, Chouinard JA, Selck C, Stertmann J, et al. Using pancreas tissue slices for in situ studies of islet of Langerhans and acinar cell biology. Nat Protoc. 2014;9(12):2809–22.
Gianani R, Putnam A, Still T, Yu L, Miao D, Gill RG, et al. Initial results of screening of nondiabetic organ donors for expression of islet autoantibodies. J Clin Endocrinol Metab. 2006;91(5):1855–61.
In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, et al. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes. 2007;56(9):2400–4.
Wiberg A, Granstam A, Ingvast S, Harkonen T, Knip M, Korsgren O, et al. Characterization of human organ donors testing positive for type 1 diabetes-associated autoantibodies. Clin Exp Immunol. 2015;182(3):278–88.
Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, et al. Increase in pancreatic proinsulin and preservation of beta cell mass in autoantibody positive donors prior to type 1 diabetes onset. Diabetes. 2017;30
Rodriguez-Calvo T, Suwandi JS, Amirian N, Zapardiel-Gonzalo J, Anquetil F, Sabouri S, et al. Heterogeneity and lobularity of pancreatic pathology in type 1 diabetes during the prediabetic phase. J Histochem Cytochem. 2015;63(8):626–36.
Mirmira RG, Sims EK, Syed F, Evans-Molina C. Biomarkers of beta-cell stress and death in type 1 diabetes. Curr Diab Rep. 2016;16(10):95.
Roder ME, Knip M, Hartling SG, Karjalainen J, Akerblom HK, Binder C. Disproportionately elevated proinsulin levels precede the onset of insulin-dependent diabetes mellitus in siblings with low first phase insulin responses. The childhood diabetes in Finland study group. J Clin Endocrinol Metab. 1994;79(6):1570–5.
Tsai EB, Sherry NA, Palmer JP, Herold KC. The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia. 2006;49:261–70.
Sherry NA, Tsai EB, Herold KC. Natural history of {beta}-cell function in type 1 diabetes. Diabetes. 2005;54(Suppl 2):S32–9.
Greenbaum CJ, Anderson AM, Dolan LM, Mayer-Davis EJ, Dabelea D, Imperatore G, et al. Preservation of beta-cell function in autoantibody-positive youth with diabetes. Diabetes Care. 2009;32(10):1839–44.
Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, et al. Improvement in outcomes of clinical islet transplantation: 1999–2010. Diabetes Care. 2012;35(7):1436–45.
Sherr JL, Ghazi T, Wurtz A, Rink L, Herold KC. Characterization of residual beta cell function in long-standing type 1 diabetes. Diabetes Metab Res Rev. 2014;30(2):154–62.
Oram RA, Jones AG, Besser RE, Knight BA, Shields BM, Brown RJ, et al. The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia. 2014;57(1):187–91.
Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, et al. Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59(11):2846–53.
Wang L, Lovejoy NF, Faustman DL. Persistence of prolonged C-peptide production in type 1 diabetes as measured with an ultrasensitive C-peptide assay. Diabetes Care. 2012;35(3):465–70.
Greenbaum CJ, Beam CA, Boulware D, Gitelman SE, Gottlieb PA, Herold KC, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite type 1 diabetes TrialNet data. Diabetes. 2012;61(8):2066–73.
Oram RA, McDonald TJ, Shields BM, Hudson MM, Shepherd MH, Hammersley S, et al. Most people with long-duration type 1 diabetes in a large population-based study are insulin microsecretors. Diabetes Care. 2015;38(2):323–8.
Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, et al. Modifying enzymes are elicited by ER stress, generating epitopes that are selectively recognized by CD4+ T cells in patients with type 1 diabetes. Diabetes. 2018;13
Acknowledgments
We would like to acknowledge Dr. Pia Leete (University of Exeter, UK) for providing immunofluorescence images. We are pleased to acknowledge financial support from the European Union’s Seventh Framework Programme PEVNET (FP7/2007–2013) under grant agreement number 261441. The participants of the PEVNET consortium are described at http://www.uta.fi/med/pevnet/publications.html. Additional support was from a Diabetes Research Wellness Foundation Non-Clinical Research Fellowship and, since 2014, a JDRF Career Development Award (5-CDA-2014-221-A-N) to S.J.R., a JDRF research grant awarded to the nPOD-V consortium (JDRF 25-2012-516), which also supports T.R.-C. and A.P. Research reviewed here involves patients from the EADB, DiViD, and nPOD collections; nPOD, The Network for Pancreatic Organ Donors with Diabetes, a collaborative type 1 diabetes research project. nPOD and A.P. are supported by grants from JDRF (5-SRA-2018-557-Q-R) and The Leona M. and Barry B. Helmsley Charitable Trust (2015PG-T1D052 and 2018PG-T1D060). Organ Procurement Organizations (OPO) partnering with nPOD to provide research resources are listed at www.jdrfnpod.org/our-partners.php.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
T.R.-C., S.J.R., and A.P. declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Studies reviewed in this article involved organ donors or deceased patients (not considered human subjects from the regulatory point of view), and living patients. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed in the animal studies reviewed in this article.
Additional information
This article is part of the Topical Collection on Pathogenesis of Type 1 Diabetes
Rights and permissions
About this article

Cite this article
Rodriguez-Calvo, T., Richardson, S.J. & Pugliese, A. Pancreas Pathology During the Natural History of Type 1 Diabetes. Curr Diab Rep 18, 124 (2018). https://doi.org/10.1007/s11892-018-1084-3
Published:
DOI: https://doi.org/10.1007/s11892-018-1084-3