
Abstract
Type 1 diabetes (T1D) is a complex autoimmune disease, and first stages of the disease typically develop early in life. Genetic as well as environmental factors are thought to contribute to the risk of developing autoimmunity against pancreatic beta cells. Several environmental factors, such as breastfeeding or early introduction of solid food, have been associated with increased risk for developing T1D. During the first years of life, the gut microbial community is shaped by the environment, in particular by dietary factors. Moreover, the gut microbiome has been described for its role in shaping the immune system early in life and early data suggest associations between T1D risk and alterations in gut microbial communities. In this article, we discuss environmental factors influencing the colonization process of the gut microbial community. Furthermore, we review possible interactions between the microbiome and the host that might contribute to the risk of developing T1D.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease being preceded with the development of autoantibodies against pancreatic beta cells. Islet autoimmunity typically occurs very early in life. Prospective cohorts with high risk individuals showed a peak at around 9 months of age [1•]. While approximately 60 % of T1D risk has been attributed to genetic factors [2], environmental risk factors such as diet, early infections, or mode of delivery are thought to significantly contribute to the risk of developing the disease [3]. Interestingly, major priming of the immune system falls within the same time frame, and factors shaping immune maturation during this period are therefore likely to contribute to the development of autoimmune disorders.
The human gastrointestinal tract constitutes a body site being equipped with a particularly high density of immune cells. Providing a major interface to the plethora of microorganisms colonizing the human gut, the role of microorganisms in priming of the immune system and their contribution to immune homeostasis has gained much interest [4]. Gut microbial colonization starts at birth and is strongly influenced by several environmental factors [5] that have also been related to autoimmunity. Apart from providing direct challenges to the immune system, the gut microbial community further interacts with the host organism through complementing its metabolism with capabilities to ferment unabsorbed nutrients, partnering the host immune system in colonization resistance, and by providing barrier against pathogenic attacks [6•]. Thus, alterations in gut microbial community composition in early infancy potentially influence the autoimmune process and pathogenesis of T1D. Indeed, early data suggest correlations between altered gut microbial communities and T1D [7–16]. However, human studies have not yet provided a clear picture and concise mechanistic explanations of the causal relationship between the gut microbiome and pathogenesis of T1D are currently lacking. One of the first studies analyzed the gut microbiome of eight children and reported a significant shift in the ratio of Firmicutes and Bacteroidetes comparing autoantibody positive and negative children [10]. In a follow-up study, Brown et al. showed that healthy children had significantly higher numbers in butyrate-producing bacteria and mucin-degrading species [7]. Further evidence of a decreased potential to produce butyrate in autoantibody-positive children has been provided by de Goffau et al. [15]. Recently, data from the Finnish DIPP cohort suggested increased abundances of Bacteroides dorei prior to autoantibody seroconversion [8]. In contrast, differences between autoantibody-positive and negative children with respect to individual microbial taxa or microbial diversity could not be detected in the German BABYDIET cohort [9]. It rather turned out that properties of microbial interaction networks are compromised in children who later developed autoantibodies [9]. Moreover, a recent follow-up study used a strategy to stratify children based on communities derived from microbial co-occurrence networks [11]. The authors suggested a functional hypothesis relating early autoantibody development with a subgroup of children that was characterized by increased Bacteroidetes and decreased Akkermansia abundances in combination with a decreased potential for the production of butyrate via the co-fermentation of acetate [11]. A recent study including 33 children from Finland and Estonia reported a drop in diversity for T1D cases after seroconversion to autoimmunity [12]. Moreover, several studies indicate increased abundances of Bacteroides species after multiple autoantibody development or T1D onset [13–16]. Although these early results suggest associations between T1D risk and alterations in the gut microbial community, most human studies so far do not provide functional models for causal associations with disease development. One reason lies in the complexity of the wealth of interactions between environment, gut microbiome, and the host organism. In this review, we summarize our current understanding of environmental factors shaping the gut microbial community in early infancy, the role of the gut microbiome in partnering the host immune system, and possible links to the development of autoimmunity and T1D.
Microbial Waves of Succession
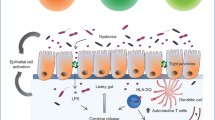
The infant gut microbiome undergoes several waves of ecological succession that are strongly influenced by factors such as host genetics, mode of delivery, and diet. Further cofactors are hygiene, social status, and treatment with drugs, in particular antibiotics (see [17] for a detailed review). The gut microbiome in early infancy is characterized by low species richness [18, 19] and is comparatively unstable [5]. Initial colonization of the ecosystem mainly occurs at birth (Fig. 1). Although recently published data indicates that the first encounter with microbial products already occurs in utero [20], mode of delivery seems to show longer lasting impact on the initial colonization of the infant gut. While vaginally delivered babies harbor a microbiome that more closely resembles the vaginal microbiome of the mother [21], infants delivered via C-section host gut microbiome more similar to the mother’s skin microbiome [21].

Microbial waves of succession. Initial colonization occurs shortly after birth. While first colonizers of vaginally delivered babies are related to the vaginal microbiome of the mother, children delivered via C-section host gut microbial communities that are more similar to the mother’s skin microbiome. In the first weeks of life, the gut microbial community is strongly influenced by breast or formula milk feeding. At the time of weaning, the variety of different glycan sources increases rapidly and the gut microbial community shifts towards and adult like community. The timing of the successional waves depends strongly on the patterns of introduction of dietary components. Here, we provide estimates which can vary strongly between individuals
Following the sequence of introduction of various food components, the gut microbial community embarks towards an adult-like community composition being characterized by increased species richness [18, 19] and higher stability [5] (Fig. 1). From the point of view of the microorganisms, initial availability of oxygen imposes a major selective pressure on the first colonizers of the neonatal gut. Therefore, aerobic or facultative aerobic species constitute the group of early colonizers (e.g., Enterobactericeae, Staphylococcus). Decreasing oxygen levels change the environment in favor of strictly anaerobic bacteria (e.g., Bifidobacterium, Clostridium, Eubacterium, and Bacteroides) [17–19, 21].
The first wave of succession is then determined by the initial diet the baby is fed with (Fig. 1). Breast milk provides a substantial source of bacteria such as Bifidobacterium, Staphylococcus, Lactococcus, Enterococcus, and Lactobacillus [22–24]. Interestingly, several of the bacteria being detected in breast milk are also being discussed for their probiotic potential [25]. High levels of Bifidobacterium spp. are commonly found in the gut of infants [26–28]. While early studies detected increased amounts of Bifidobacterium in breast-fed infants compared to bottle-fed infants, more recent studies do not confirm such differences (see [17] for a detailed review). Apart from technical issues such as the availability of suitable primers [19], it has been argued that such controversial results might follow from an increased similarity between human breast milk and modern formula milk being enriched with prebiotics (e.g., galactooligosaccharides and fructooligosaccharides) [17, 29–31]. Indeed, a comparison of different brands of formula showed that enrichment of formula milk with oligosaccharides can be associated with increased abundances of Bifidobacterium and Lactobacillus in stool samples [32]. Human breast milk contains large amounts of human milk oligosaccharides (HMOs) which show high similarity to O- and N-linked gylcans being part of mucin proteins found in the intestinal mucus layer [33••]. As HMOs cannot be dissected by human enzymes, they might provide a competitive advantage [33••] for specialized bacteria that are able to use HMOs or their components as sources of energy (e.g., Bifidobacterium [34–37] or Lactobacillus [38, 39]). Taking into account that a significant portion of the energy provided by human breast milk cannot be absorbed by the infant, it is assumed that the similarity of HMOs with mucus-derived glycans results from co-evolution of humans with intestinal commensals, preparing the gut ecosystem for the later colonization with mucus degrading bacteria [33••].
After weaning, complex carbohydrates such as cereals, fruits, and vegetables enrich infant diet and the variety of glycan sources increases rapidly [33••] (Fig. 1). Accordingly, the composition of the gut microbiome shifts from typical utilizers of HMOs towards bacteria that are able to ferment other dietary components managing to pass the gastro-intestinal passage. Typically, abundances of Firmicutes, Bacteroidetes, and Actinobacteria increase while Bifidobacterium, Enterobacteriaceae, and lactic acid bacteria decrease during this period [28]. Composition and sequence of introduction of dietary compounds further constitute primary factors of community composition. While generalists are able to feed on various dietary sources, the abundance of specialists strongly rely on the availability of the special type of host diet providing their favorite substrate. To survive over longer periods, specialists must have a constant supply with their preferred energy source. They will therefore only persist, if the latter is constantly available [33••]. One prominent example is the mucus degrading specialist Akkermansia muciniphila, which is characterized by a high number of genes involved in the degradation of human-derived glycans [40]. In contrast, generalists such as Bacteroides thetaiotaomicron may shift their metabolism towards fermentation of luminal content, depending on other dietary sources being available [33••]. In addition to the shift in microbial community composition due to the transition from breast milk to a more complex diet, short-term fluctuations in nutrients on a meal-to-meal basis can also affect the microbial population [41–43]. Moreover, different processing of dietary components (e.g., cooking, milling) can potentially influence microbial composition as, e.g., dietary starch changes its chemical properties depending on the type of processing [44].
Apart from any direct modulation of the intestinal community composition via dietary compounds, there are many indirect effects due to competition and facilitation within the bacterial ecosystem. For example, B. thetaiotaomicron consumes carbohydrates which are otherwise used by certain Clostridium species, thus executing competitive exclusion [45]. Direct colonization resistance is realized through the production of bacteriocins, targeting competitors via impairing growth or adhesion [45, 46]. Other mechanisms utilize the host immune system, such as the capability of B. thetaiotaomicron to enhance the expression of genes which lead to an increased production of antimicrobial peptides [45]. Less targeted drivers of community composition are given by a variety of cross-feeding effects which, for example, are thought to determine the distribution of short-chain fatty acid (SCFA) production to a large extent [47]. Successful niching therefore typically results from matching the species’ metabolic repertoire with the availability of energy sources as well as the ability to modulate environmental factors towards favorable conditions. Although major drivers of community composition are known, sensitivity and robustness of the complex network of microbial interactions need to be better understood. On top of that, it has to be expected that many more interactions and interrelations are yet to be discovered.
Host Microbiome Interactions
Many commensal gut microbiota co-evolved with their host organisms and thus have developed specialized mechanisms of interaction with their human environment. Likewise, human metabolism and immune system rely in multiple ways on the presence of microbial partners. The most obvious contribution of the gut microorganisms is fermentation of non-soluble dietary compounds, which cannot be absorbed by the host without prior degradation. Products of microbial fermentation significantly contribute to the energy intake of the host. Apart from being an additional source of energy, end products of microbial metabolism also serve other health-promoting functions. For example, SCFAs have been shown to modulate immune function [48, 49••, 50, 51]. On the other side, microbial-associated molecular patterns (MAMP) provide an early immunological challenge, guiding the human immune system from the first moment of contact on.
Shortly after birth, the immune system is considered to be relatively immature, enabling the colonization of the gut with commensal microbiota from the environment (see [4] for a detailed review). During the development of the host’s immune system, MAMPs play a central role in learning to maintain a healthy balance between pro- and anti-inflammatory immune reaction [4, 52]. Apart from modulatory function, gut microbes interfere with colonization processes through mechanisms of colonization resistance, such as direct inhibition via bacteriocins, competition for nutrients or receptors, or direct barrier function via building of biofilms.
On the other side, the host protects itself from microbial invasion. There are several immune mechanisms confining the gut microbiota to the lumen. A first, physical barrier is provided by a mucus layer, consisting of a variety of glycoproteins secreted by goblet cells. In the large intestine, this layer consists of an inner and an outer mucus layer [53]. While the latter provides an attachment site and endogenous nutrient source for commensal bacteria [54], the inner layer is commonly not colonized by bacterial populations, thus providing a dense physical barrier against bacterial penetration of the epithelium [53]. There is increasing evidence suggesting that structure and composition of the mucosal barrier result from host-microbial interaction. For instance, a recent study showed that, in contrast to an intact mucus layer in conventionally raised mice, the inner layer of germ-free mice can be penetrated by bacteria-sized particles. After colonization of germ-free mice with microbiota from conventionally raised animals, mucus function has been recovered after 6 weeks [55].
In contrast to the large intestine, the mucosal surface in the small intestine consists of one layer of mucus, only, and host-derived antimicrobial peptides (AMPs) are heavily utilized to prevent the epithelium from microbial penetration [56]. Paneth cells located in the epithelium in the small intestine are an important source of AMPs [57]. Microbes can be sensed by Paneth cells in a MyD88-dependent manner, resulting in the production of AMPs (e.g., defensins and C-type lectins) [58]. The latter includes the C-type lectin RegIII-γ which inhibits adhesion of Gram-positve bacteria to epithelial cells [59]. Furthermore, defensins such as α-defensin shape the microbial community by directly targeting the bacterial cell wall or by binding to the microbial cell surface (see [56] for a detailed review). Thus, AMPs contribute to the regulation of the composition of the bacterial community besides preventing the epithelium from microbial encounter.
Another strategy to confine gut microbiota to the lumen relies on secreted immunoglobulin A (IgA). MAMPs are sensed by dendritic cells which, as a consequence, stimulate the production of IgA by B cells originating from Peyer’s patches [60]. Secreted IgA then is transcytosed into the lumen and protects further bacterial translocation through the epithelium by binding to microbial antigens [60]. Thus, being a first line of defense, IgA plays a key role in maintaining integrity of the epithelial barrier and thus prevents systemic immune responses resulting from breaches of the gut epithelium. Interestingly, experiments in mice indicate that alterations in the specificity of the IgA repertoire can lead to substantial shifts in microbial community composition [61].
In addition to the production of IgA and AMPs, gut microbiota substantially alter immune homeostasis by impacting the differentiation of immune cell subsets. A fine-tuned balance between pro- and anti-inflammatory immune responses is crucial to remain tolerogenic for commensals and, at the same time, being able to defend against invasion by pathogens. With respect to the development of autoimmunity, any excess in pro-inflammatory immune responses is clearly not desirable. As one line of evidence in this direction, increased Th17 (IL-17) responses have been associated with the development of autoimmune diseases [62, 63]. On the other hand, pro-inflammatory Th17 responses are necessary to prevent pathogenic infections, suggesting that a balanced degree of Th17 response is crucial for immune homeostasis [64]. Another concrete mechanism has been elucidated in the mouse model where segmented filamentous bacteria (SFB) have been shown to induce pro-inflammatory responses due to the differentiation of Th17 cells [62, 65, 66]. A similar effect has been shown via the production of adenosine 5′-triphosphate (ATP) by commensal bacteria [67].
On the other hand, commensal gut microbiota can also induce anti-inflammatory effects, for instance by the suppression of Th17 via regulatory T cells (Treg). One prominent example is the human commensal Bacteroides fragilis which induces differentiation of Foxp3+ Tregs from CD4+ T cells. This conversion is mediated by the MAMP polysaccharide A (PSA) from the outer membrane of B. fragilis [68]. Similarly, colonization of germ-free mice with bacterial strains from the Clostridium clusters IV and XIVa, or with bacteria from the altered Schaedler flora (ASF) resulted in the differentiation of Tregs in the lamina propria [69, 70]. Most Treg cells are selected in the thymus and suppress T cell subsets associated with autoimmune reactions [64]. However, induction of Treg cells from naïve T cells by gut microbiota likely occurs post-thymic in the periphery of the colon (see [71] for a detailed review). Supporting this line of thought, germ-free mice have been found to harbor reduced amounts of peripherally derived Treg cells (pTreg), suggesting that commensal microbiota drive pTreg cell generation [69, 70, 72].
Several studies suggest that end products of bacterial metabolism, such as SCFAs, provide a potential route by which commensals contribute to enhanced generation of pTreg cells [49••, 50, 51]. Interestingly, among SCFAs, butyrate has been found to have the most significant contribution to colonic pTreg induction [49••, 50]. In addition, Chang et al. recently demonstrated that butyrate shows anti-inflammatory effects by down-regulating macrophage-induced pro-inflammatory responses [48]. Apart from SCFAs, other microbial-derived products such as bile acids, vitamins, amino acids, and sphingolipids can also substantially influence immune homeostasis [73, 74].
Butyrate does not only impact the generation of regulatory T cells but also has several other health-promoting effects for the host organism. For instance, butyrate is considered to be the main energy source for intestinal epithelial cells [76] and enhances mucus production [76, 77]. Thus, in addition to its immune regulatory potential, increased butyrate production can have a profound impact on the integrity of the gut barrier. Production of SCFAs results from microbial fermentation of dietary glycans. In addition, endogenously derived glycans such as mucins can provide further sources. As outlined above, composition of dietary compounds strongly influences microbial community composition as various bacteria have different strategies to utilize different types of glycans (see [33] for a detailed review).
For a detailed understanding of the interplay of various mechanisms of butyrate production in the intestinal microbial community, cross-feeding effects need to be considered. Co-fermentation experiments have documented butyrogenic effects in multiple bacterial compositions. A typical example is an enhanced butyrate production due to cross-feeding of Bifidobacterium adolescentis and the lactate-utilizing butyrate producers Eubacterium hallii and Anaerostipes caccae [78]. Moreover, some butyrate-producing species such as Roseburia spp. and Faecalibacterium spp. require acetate for their growth [79, 80], and bacteria-producing acetate therefore exhibit a significant impact on butyrate production.
Two different pathways have been described for the production of butyrate from butyryl-CoA. Compared to the classical buk-pathway (phosphotransbutyrylase and butyrate-kinase via butyryl-phosphate), the recently described but-pathway (butyryl-CoA: acetate CoA-transferase) utilizes acetate as a co-substrate for butyrate synthesis. Interestingly, the but-pathway is far more prevalent among butyrate-producing species [81]. In the presence of acetate, the net output of butyrate from the but-pathway is higher than with butyrate production via the buk-pathway [79]. Hence, increased availability of acetate is a critical driver for butyrate production. Fermentation of butyrate results in the production of hydrogen (H2) which has to be removed in order to not inhibit further butyrate production [82]. Consequently, hydrogen utilization by hydrogenotrophic bacteria has been shown to strongly influence SCFA production [83]. Acetate is a fermentation product of most enteric bacteria and is also produced by acetogens by utilizing H2 and CO2 [84]. Acetogens compete with other hydrogenotrophic bacteria (methanogens and sulfate-reducing bacteria (SRBs)) for the available H2. In the presence of sulfate, acetogens and methanogens are outcompeted by SRBs [85]. In contrast, however, lower levels of sulfate lead to increased abundances of acetogens [85] which might enhance butyrate production via co-fermentation of acetate. One possible driver of sulfate availability might lie in the competition for mucins by various mucin-degrading species. With mucins being a major source of endogenously derived sulfate, variability in sulfate utilization likely results in compositional changes of the microbial community. While most mucin-degrading bacteria are not able to fully degrade mucins and therefore release sulfate [86], other specialists such as A. muciniphila are able to use sulfate in an assimilatory manner [87]. Lower levels of sulfate might then promote a butyrogenic effect through increased availability of acetate provided by acetogenic bacteria.
Possible Impacts on the Pathogenesis of T1D
In patients developing T1D, T cell-mediated autoimmune reactions lead to the destruction of pancreatic beta cells in the islet of Langerhans. There is a close immunological link between the gastrointestinal tract and the pancreas as antigens derived from dietary compounds or gut microbiota might impair pancreatic immune response (see [88] for a recent review). Several models have been proposed linking the gut microbiome with the development of T1D. These include the Leaky Gut Hypothesis, the Hygiene Hypothesis, the Old Friends Hypothesis, and the Perfect Storm Hypothesis. According to the Leaky Gut Hypothesis, increased permeability of the gut epithelium results from loss of tight barrier function [89]. Thus, diet-derived macro-molecules and microbial antigens can pass the epithelial barrier and consequently trigger intestinal inflammation. Further downstream, this might possibly lead to pancreatic beta cell attack [88]. Indeed, several studies indicate that individuals with T1D show increased permeability of the gut epithelium [90–94]. Impaired butyrate production provides one explanation for a loss of tight barrier function since butyrate is known to be the major source of energy for gut epithelial cells [75].
On a somewhat more general basis, the Old Friends Hypothesis stresses the role of commensal gut microbiota having co-evolved with their host organisms over long periods of time. Therefore, loss of contact with these microbial partners may show significant effects on the ability of the host’s immune system in educating its regulatory arm and maintaining homeostasis [95]. In healthy individuals, autoreactive immune reactions can be suppressed by Tregs generated in the thymus as well as in the periphery. Thus, lack of encounter with co-evolved commensal bacteria might substantially influence self/non-self-recognition patterns.
The Perfect Storm Hypothesis combines ideas from the Leaky Gut Hypothesis and the Old Friends Hypothesis by suggesting that a combination of increased permeability of the epithelial barrier, an altered microbial community composition, and an impaired intestinal immune responsiveness interact collectively, leading to the development of anti-islet autoimmunity [96].
Along the lines of the Old Friends Hypothesis, the Hygiene Hypotheses claims that increasing T1D incidences being observed in Western societies result from a lack of contact with infectious agents due to increased hygienic conditions (see [97] for a recent review). Missing out pathogenic encounter in early childhood, the immune system lacks important early challenges which are required for proper priming. On the long run, such impairment leads to over-reaction of the immune system resulting in autoimmunity.
A key for deriving a mechanistic explanation for the described hypotheses might lie in shifts in the SCFA balance, especially regarding butyrate production. Support for this claim comes from several cohort studies, showing associations between autoimmune diseases and reduced butyrate levels [7, 11, 15]. Taken together, the gut microbiome has to be considered being a key factor in the early development of the host’s immune system. Therefore, a detailed understanding of the functional interplay between the gut microbiome and the host will possibly provide valuable insights into some of the environmental factors driving autoimmunity early in life.
The development of anti-islet cell autoantibodies against pancreatic beta cells nicely fits into this scheme. With autoimmunity peaking around 9 months of age [1], this period coincides with both substantial shifts in infant diet and with early priming of the immune system. Being the major driver of microbial community composition in the gut, dietary factors are expected to contribute to functional associations between gut microbiome and development of the disease. Opposition might stem from controversially discussed observations, such as the association of breast feeding with T1D development [98–100]. Nevertheless, heterogeneity of microbiome composition might well provide a basis for seemingly opposing observations. For example, differences in the type of formula milk being provided might as well significantly contribute to the observed heterogeneity as might other social and geographical factors leading to variations in the composition of the intestinal microbial community. Taking into account that the composition of formula milk constantly changes based on new scientific insight and further differs between geographic locations, the impact of formula diet on the gut microbial community is highly variable [32]. Furthermore, early introduction of solid food components, such as cereals, fruits, and berries, have been identified as possible factors contributing to the development of the disease [98, 101, 102]. Again, these dietary factors do not only vary both geographically and with social status [103], but can also lead to substantial shifts in the microbial community [104]. One line of argumentation is based on the exact compounds being used to replace HMOs in industrially produced formula [105]. Accepting the position that HMOs provide an evolutionary derived competitive advantage for mucin-degrading specialists such as A. muciniphila, early introduction of additional glycan sources might well shift the microbial community, favoring generalists (e.g., Bacteroides spp.) rather than the co-evolved specialists [11]. To understand the role of the gut microbiome in T1D, it will be crucial to combine the analysis of dietary patterns with a better understanding on factors driving microbial community composition in the gut and its consequences for human metabolism and early development of the immune system [106].
In addition to dietary factors, microbial community composition in the infant gut is further influenced by other co-factors such as mode of delivery, drug treatment, or other factors, such as geographic location. Interestingly, several of those have been associated with an increased risk of development of T1D. Thus, the microbiome provides a suitable route for understanding such relations and, at the same time, explaining controversial findings. For instance, highest incidences of T1D have been reported in Finland [107], and recent results from the TEDDY cohort indicate that Finnish children show relatively low bacterial diversity and increased abundances of Bacteroides spp. in combination with decreased abundances of Akkermansia [108]. Thus, geographical differences in microbial community composition might explain the observed differences in T1D incidences.
Along these lines, birth via Caesarean section has been associated with increased risk for T1D development [109, 110]. Several hypotheses have been discussed to explain a possible association of T1D and mode of delivery [111]. One possible explanation is that vaginally delivered babies obtain their initial microbiome from the vaginal microbiome of their mothers [21]. Hence, the initial microbial colonization with commensal and/or pathogenic microbiota might contribute in shaping the initial immune system. Again, non-conclusive observations might be explained with variations in the stage of dietary maturation of the children, which in most cases has not been accessed. Even more, a combination of altered gut microbial community composition coming along with certain genetic factors, such as the IFIH1 polymorphism, can potentially enhance the risk for T1D in children delivered by Caesarean section [110]. Other genetic risk factors which also show a link to altered microbial colonization are HLA genotype [112] and secretor state FUT2 [113, 114]. Several other genes associated with the host immune system have also been described for their impact on gut microbial composition [5].
Conclusions and Future Directions
Due to significant improvements in sequencing technologies, we are now more and more able to access the full diversity of the human gut microbial community and characterize its metabolic potential. However, with regard to T1D, we are just beginning to understand associations between the gut microbiome and pathogenesis of the disease. Factors contributing to disease development might be highly heterogeneous between individuals and it is likely that a combination of diverse factors is responsible for autoimmunity development. To tackle the complexity and the heterogeneity of this disease, large sample size cohorts and approaches integrating data from various sources will be required in future. Data from the TEDDY cohort [115] is currently analyzed, aiming at the identification of various factors contributing to disease development. The amount of different data sources being provided, the dense longitudinal sampling starting shortly after birth, as well as the large number of individuals being included in this cohort, provide a unique opportunity to tackle the complexity of these questions.
Recent advances in sequencing technologies revealed the comprehensive significance of viruses in shaping the functional repertoire of bacterial associations. Notwithstanding being important, this aspect has deliberately been left out of discussion for this review. It well deserves consideration on its own. For instance, bacteriophages substantially shape the microbial community and most likely also contribute to dysbiosis (see [116] for a recent review). The role of bacteriophages in shaping the infant immune system has rarely been studied. Phages might influence the immune system either directly or by modifying bacterial functions [117]. Thus, the complexity of these associations provides an additional level of complexity for analysis.
To understand microbial factors contributing to disease development, it will be crucial to unravel environmental factors shaping microbial community composition early in life and its impact on the developing immune system. Among these microbial factors, the potential to produce various fermentation products is strongly influenced by the diet of the host. Early data suggest that alterations in the proportions of SCFAs, such as butyrate, might be a potential mechanism contributing to the risk of developing T1D [7, 11, 15]. Since dietary changes strongly shift microbial community composition early in life, it will be of substantial importance for the success of the endeavor to include detailed dietary information into the analyses focusing on the association of the gut microbiome and T1D development [11]. Furthermore, starting with birth, early gut microbial communities undergo several changes triggered by the environment. Thus, very early sampling of the microbiome might be required to unravel the functional impact of the microbial community with regard to the developing infant immune system. As the gut microbiome constitutes a full ecosystem with microbial species depending on each other and competing for nutrient sources, it might not be sufficient to focus on individual microbes only, but rather to focus on interaction patterns of the community as a whole. The latter bears the chance to unravel functional relationships regarding the complex system of host microbiota interactions with the potential towards future strategies for prevention.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Krischer JP, Lynch KF, Schatz DA, et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia. 2015;58(5):980–7. Interesting data from the TEDDY study, showing the incidence of autoantibody development over time.
Eringsmark Regnell S, Lernmark A. The environment and the origins of islet autoimmunity and Type 1 diabetes. Diabet Med. 2013;30(2):155–60.
Knip M, Simell O. Environmental triggers of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(7):a007690.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–41.
Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9(4):279–90.
Sommer F, Backhed F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–38. Useful review article discussing the role of the gut microbiome in the development of the host immune system, metabolsim and physiology.
Brown CT, Davis-Richardson AG, Giongo A, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6(10):e25792.
Davis-Richardson AG, Ardissone AN, Dias R, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. 2014;5:678.
Endesfelder D, zu Castell W, Ardissone A, et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes. 2014;63(6):2006–14.
Giongo A, Gano KA, Crabb DB, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91.
Endesfelder D, Engel M, Davis-Richardson AG, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. 2016; 4(1):17.
Kostic AD, Gevers D, Siljander H, et al. The dynamics of the human infant gut microbiome in development and in progression toward Type 1 diabetes. Cell Host Microbe. 2015;17(2):260–73.
Murri M, Leiva I, Gomez-Zumaquero JM, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case–control study. BMC Med. 2013;11:46.
Mejia-Leon ME, Petrosino JF, Ajami NJ, et al. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci Rep. 2014;4:3814.
de Goffau MC, Luopajarvi K, Knip M, et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. 2013;62(4):1238–44.
Alkanani AK, Hara N, Gottlieb PA, et al. Alterations in intestinal microbiota correlate with susceptibility to Type 1 diabetes. Diabetes. 2015;64(10):3510–20.
Adlerberth I, Wold AE. Establishment of the gut microbiota in Western infants. Acta Paediatr. 2009;98(2):229–38.
Koenig JE, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4578–85.
Palmer C, Bik EM, DiGiulio DB, et al. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5(7):e177.
Aagaard K, Ma J, Antony KM, et al. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65.
Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–5.
Martin R, Langa S, Reviriego C, et al. Human milk is a source of lactic acid bacteria for the infant gut. J Pediatr. 2003;143(6):754–8.
Kawada M, Okuzumi K, Hitomi S, et al. Transmission of Staphylococcus aureus between healthy, lactating mothers and their infants by breastfeeding. J Hum Lact. 2003;19(4):411–7.
Gronlund MM, Lehtonen OP, Eerola E, et al. Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J Pediatr Gastroenterol Nutr. 1999;28(1):19–25.
Lara-Villoslada F, Olivares M, Sierra S, et al. Beneficial effects of probiotic bacteria isolated from breast milk. Br J Nutr. 2007;98 Suppl 1:S96–100.
Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, et al. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30(1):61–7.
Turroni F, Peano C, Pass DA, et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One. 2012;7(5):e36957.
Bergstrom A, Skov TH, Bahl MI, et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol. 2014;80(9):2889–900.
Marques TM, Wall R, Ross RP, et al. Programming infant gut microbiota: influence of dietary and environmental factors. Curr Opin Biotechnol. 2010;21(2):149–56.
Boehm G, Moro G. Structural and functional aspects of prebiotics used in infant nutrition. J Nutr. 2008;138(9):1818S–28S.
Oozeer R, van Limpt K, Ludwig T, et al. Intestinal microbiology in early life: specific prebiotics can have similar functionalities as human-milk oligosaccharides. Am J Clin Nutr. 2013;98(2):561S–71S.
Penders J, Thijs C, Vink C, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118(2):511–21.
Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10(5):323–35. This is an Important review article that discusses several factors how differences in glycan availability influence the gut microbial community.
Marcobal A, Barboza M, Sonnenburg ED, et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe. 2011;10(5):507–14.
Miwa M, Horimoto T, Kiyohara M, et al. Cooperation of beta-galactosidase and beta-N-acetylhexosaminidase from bifidobacteria in assimilation of human milk oligosaccharides with type 2 structure. Glycobiology. 2010;20(11):1402–9.
LoCascio RG, Desai P, Sela DA, et al. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl Environ Microbiol. 2010;76(22):7373–81.
Sela DA. Bifidobacterial utilization of human milk oligosaccharides. Int J Food Microbiol. 2011;149(1):58–64.
Schwab C, Ganzle M. Lactic acid bacteria fermentation of human milk oligosaccharide components, human milk oligosaccharides and galactooligosaccharides. FEMS Microbiol Lett. 2011;315(2):141–8.
Martinez RC, Aynaou AE, Albrecht S, et al. In vitro evaluation of gastrointestinal survival of Lactobacillus amylovorus DSM 16698 alone and combined with galactooligosaccharides, milk and/or Bifidobacterium animalis subsp. lactis Bb-12. Int J Food Microbiol. 2011;149(2):152–8.
van Passel MW, Kant R, Zoetendal EG, et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS One. 2011;6(3):e16876.
Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108(15):6252–7.
Turnbaugh PJ, Ridaura VK, Faith JJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14.
Faith JJ, McNulty NP, Rey FE, et al. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science. 2011;333(6038):101–4.
Englyst HN, Kingman SM, Cummings JH. Classification and measurement of nutritionally important starch fractions. Eur J Clin Nutr. 1992;46 Suppl 2:S33–50.
Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801.
Martinez FA, Balciunas EM, Converti A, et al. Bacteriocin production by Bifidobacterium spp A review. Biotechnol Adv. 2013;31(4):482–8.
De Vuyst L, Leroy F. Cross-feeding between bifidobacteria and butyrate-producing colon bacteria explains bifdobacterial competitiveness, butyrate production, and gas production. Int J Food Microbiol. 2011;149(1):73–80.
Chang PV, Hao L, Offermanns S, et al. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–52.
Furusawa Y, Obata Y, Fukuda S. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. This important study shows that butyrate production by commensal microbiota induces the differentiation of regulatory T cells in the colonic lamina propria of mice.
Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–5.
Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–73.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23.
Johansson ME, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105(39):15064–9.
Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4659–65.
Johansson, M.E., H.E. Jakobsson, J. Holmen-Larsson, et al., Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe, 2015
Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol. 2013;14(7):660–7.
Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9(5):356–68.
Vaishnava S, Behrendt CL, Ismail AS, et al. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105(52):20858–63.
Vaishnava S, Yamamoto M, Severson KM, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–8.
Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303(5664):1662–5.
Kawamoto S, Tran TH, Maruya M, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336(6080):485–9.
Wu HJ, Ivanov II, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–27.
Lee YK, Menezes JS, Umesaki Y, et al. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4615–22.
Arrieta MC, Finlay BB. The commensal microbiota drives immune homeostasis. Front Immunol. 2012;3:33.
Ivanov II, Atarashi K, Mane N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98.
Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31(4):677–89.
Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–12.
Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107(27):12204–9.
Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–41.
Geuking MB, Cahenzli J, Lawson MA, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34(5):794–806.
Hoeppli RE, Wu D, Cook L, et al. The environment of regulatory T cell biology: cytokines, metabolites, and the microbiome. Front Immunol. 2015;6:61.
Lathrop SK, Bloom SM, Rao SM, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478(7368):250–4.
Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14(7):676–84.
An D, Oh SF, Olszak T, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156(1–2):123–33.
Hamer HM, Jonkers D, Venema K, et al. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–19.
Finnie IA, Dwarakanath AD, Taylor BA, et al. Colonic mucin synthesis is increased by sodium butyrate. Gut. 1995;36(1):93–9.
Shimotoyodome A, Meguro S, Hase T, et al. Short chain fatty acids but not lactate or succinate stimulate mucus release in the rat colon. Comp Biochem Physiol A Mol Integr Physiol. 2000;125(4):525–31.
Pryde SE, Duncan SH, Hold GL, et al. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217(2):133–9.
Duncan SH, Holtrop G, Lobley GE, et al. Contribution of acetate to butyrate formation by human faecal bacteria. Br J Nutr. 2004;91(6):915–23.
Barcenilla A, Pryde SE, Martin JC, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. 2000;66(4):1654–61.
Louis P, Duncan SH, McCrae SI, et al. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J Bacteriol. 2004;186(7):2099–106.
Flint HJ, Duncan SH, Scott KP, et al. Interactions and competition within the microbial community of the human colon: links between diet and health. Environ Microbiol. 2007;9(5):1101–11.
Chassard C, Bernalier-Donadille A. H2 and acetate transfers during xylan fermentation between a butyrate-producing xylanolytic species and hydrogenotrophic microorganisms from the human gut. FEMS Microbiol Lett. 2006;254(1):116–22.
Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12(10):661–72.
Dar SA, Kleerebezem R, Stams AJ, et al. Competition and coexistence of sulfate-reducing bacteria, acetogens and methanogens in a lab-scale anaerobic bioreactor as affected by changing substrate to sulfate ratio. Appl Microbiol Biotechnol. 2008;78(6):1045–55.
Willis CL, Cummings JH, Neale G, et al. In vitro effects of mucin fermentation on the growth of human colonic sulphate-reducing bacteria. Anaerobe. 1996;2(2):117–22.
KEGG Database, http://www.genome.jp/kegg-bin/show_pathway?amu00920. Accessed 26 November 2015.
Vaarala O. Is the origin of type 1 diabetes in the gut? Immunol Cell Biol. 2012;90(3):271–6.
Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol. 2008;24(6):701–6.
Kuitunen M, Saukkonen T, Ilonen J, et al. Intestinal permeability to mannitol and lactulose in children with type 1 diabetes with the HLA-DQB1*02 allele. Autoimmunity. 2002;35(5):365–8.
Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55(5):1443–9.
Secondulfo M, Iafusco D, Carratu R, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis. 2004;36(1):35–45.
Bosi E, Molteni L, Radaelli MG, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. 2006;49(12):2824–7.
Mooradian AD, Morley JE, Levine AS, et al. Abnormal intestinal permeability to sugars in diabetes mellitus. Diabetologia. 1986;29(4):221–4.
Rook GA, Brunet LR. Microbes, immunoregulation, and the gut. Gut. 2005;54(3):317–20.
Vaarala O, Atkinson MA, Neu J. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. 2008;57(10):2555–62.
Bach JF, Chatenoud L. The hygiene hypothesis: an explanation for the increased frequency of insulin-dependent diabetes. Cold Spring Harb Perspect Med. 2012;2(2):a007799.
Virtanen SM, Kenward MG, Erkkola M, et al. Age at introduction of new foods and advanced beta cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes. Diabetologia. 2006;49(7):1512–21.
Cardwell CR, Stene LC, Ludvigsson J, et al. Breast-feeding and childhood-onset type 1 diabetes: a pooled analysis of individual participant data from 43 observational studies. Diabetes Care. 2012;35(11):2215–25.
Ziegler AG, Schmid S, Huber D, et al. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA. 2003;290(13):1721–8.
Chmiel R, Beyerlein A, Knopff A, et al. Early infant feeding and risk of developing islet autoimmunity and type 1 diabetes. Acta Diabetol. 2015;52(3):621–4.
Norris JM, Barriga K, Klingensmith G, et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA. 2003;290(13):1713–20.
Aronsson CA, Lee HS, Liu E, et al. Age at gluten introduction and risk of celiac disease. Pediatrics. 2015;135(2):239–45.
Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–7.
Bode L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology. 2012;22(9):1147–62.
Semenkovich CF, Danska J, Darsow T, et al. American Diabetes Association and JDRF Research Symposium: diabetes and the microbiome. Diabetes. 2015;64(12):3967–77.
Karvonen M, Viik-Kajander M, Moltchanova E, et al. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 2000;23(10):1516–26.
Kemppainen KM, Ardissone AN, Davis-Richardson AG, et al. Early childhood gut microbiomes show strong geographic differences among subjects at high risk for type 1 diabetes. Diabetes Care. 2015;38(2):329–32.
Cardwell CR, Stene LC, Joner G, et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. 2008;51(5):726–35.
Bonifacio E, Warncke K, Winkler C, et al. Cesarean section and interferon-induced helicase gene polymorphisms combine to increase childhood type 1 diabetes risk. Diabetes. 2011;60(12):3300–6.
Vehik K, Dabelea D. Why are C-section deliveries linked to childhood type 1 diabetes? Diabetes. 2012;61(1):36–7.
De Palma G, Capilla A, Nadal I, et al. Interplay between human leukocyte antigen genes and the microbial colonization process of the newborn intestine. Curr Issues Mol Biol. 2010;12(1):1–10.
Rausch P, Rehman A, Kunzel S, et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108(47):19030–5.
Yang P, Li HL, Wang CY. FUT2 nonfunctional variant: a “missing link” between genes and environment in type 1 diabetes? Diabetes. 2011;60(11):2685–7.
Group TS. The Environmental Determinants of Diabetes in the Young (TEDDY) Study. Ann N Y Acad Sci. 2008;1150:1–13.
Mills S, Shanahan F, Stanton C, et al. Movers and shakers: influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes. 2013;4(1):4–16.
De Paepe M, Leclerc M, Tinsley CR, et al. Bacteriophages: an underestimated role in human and animal health? Front Cell Infect Microbiol. 2014;4:39.
Acknowledgments
David Endesfelder and Wolfgang zu Castell report grants from the Juvenile Diabetes Research Foundation. We thank Ms. Sandra Mayer for the production of Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
David Endesfelder, Marion Engel, and Wolfgang zu Castell declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Transplantation
Rights and permissions
About this article

Cite this article
Endesfelder, D., Engel, M. & zu Castell, W. Gut Immunity and Type 1 Diabetes: a Mélange of Microbes, Diet, and Host Interactions?. Curr Diab Rep 16, 60 (2016). https://doi.org/10.1007/s11892-016-0753-3
Published:
DOI: https://doi.org/10.1007/s11892-016-0753-3