
Abstract
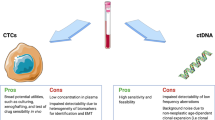
Circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) are emerging noninvasive multifunctional biomarkers in liquid biopsy allowing for early diagnosis, accurate prognosis, therapeutic target selection, spatiotemporal monitoring of metastasis, as well as monitoring response and resistance to treatment. CTCs and ctDNA are released from different tumor types at different stages and contribute complementary information for clinical decision. Although big strides have been taken in technology development for detection, isolation and characterization of CTCs and sensitive and specific detection of ctDNA, CTC-, and ctDNA-based liquid biopsies may not be widely adopted for routine cancer patient care until the suitability, accuracy, and reliability of these tests are validated and more standardized protocols are corroborated in large, independent, prospectively designed trials. This review covers CTC- and ctDNA-related technologies and their application in colorectal cancer. The promise of CTC- and ctDNA-based liquid biopsies is envisioned.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths, and nearly 50,000 people will die from this disease in the USA in 2015 [1, 2]. Half of CRC patients develop local recurrence or distant metastasis. Although traditional tissue biopsies and imaging studies remain the gold standard in metastatic cancer care, the spatiotemporal dynamic heterogeneity of cancer limits their utility. The idea of a minimally invasive way to obtain accurate information from a blood sample, also known as liquid biopsy, has gained increasing attention in cancer diagnosis, risk stratification, and monitoring treatment response. Circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) are two main biomarkers detected in liquid biopsies. Although CTCs and ctDNA were identified in 1869 and 1948, respectively, their prognostic and diagnostic applications in oncology were not realized until two decades ago with the development of various technological advancements [3, 4]. Information derived from CTCs and ctDNA offer a unique opportunity to enrich our understanding of cancer biology, tumor evolution, and therapeutic efficacy and resistance. CTCs and ctDNA as liquid biopsies are some of the newest trends in current translational cancer research with about 380 clinical trials using CTCs and/or ctDNA as biomarkers. In this review, we explore recent advances in CTC and ctDNA-related technologies and their translational applications in the diagnosis and management of CRC, as well as the pros and cons of these approaches.
CTCs
Cancer heterogeneity poses important pathological, diagnostic, and therapeutic challenges for both clinicians and scientists. Traditional tissue biopsies are invasive with associated risks and discomfort for patients; at times, they are impossible or impractical due to the tumor’s location or presence of multiple lesions. These issues make serial monitoring during treatment problematic, undesirable, and occasionally, impossible. In some cases, liquid biopsies may potentially replace or complement regular tissue biopsies to achieve accurate early detection of cancer, relapse, or progression, provide personalized treatments, and overcome challenges posed by cancer heterogeneity. CTCs are intact cancer cells emanating from a primary tumor and/or metastatic lesions that can be detected in the blood of patients with malignancies. They represent a heterogeneous pool of tumor cells. The presence of CTCs is a strong and independent prognostic marker in patients with several major cancer types including CRC in both the metastatic and non-metastatic setting [5•]. The identification and analysis of CTCs may potentially aid in early and accurate cancer diagnosis, detection of metastases or relapse, evaluation of treatment efficacy, monitoring for the emergence of treatment resistance, and identification of new therapeutic targets to guide management. Early detection of resistant cancer cells is important because they have the ability to metastasize aggressively.
In 2004, the US Food and Drug Administration (FDA) approved the CellSearch system (Veridex LLC, Raritan, NJ) as a method for CTC detection following multiple studies establishing CTCs as an independent prognostic marker in metastatic breast cancer, castration-resistant prostate cancer, and metastastic CRC (mCRC) [6–8]. However, CTC enumeration alone provides limited information. Further characterization with phenotypic and genomic analyses of CTCs provides unprecedented insights into the biology of metastasis and mechanisms of treatment resistance, information on biomarkers predictive of drug sensitivity, and identification of therapeutic targets. Despite a multitude of technological advancements, we continue to face new challenges when studying CTCs. In-depth investigation is limited by low numbers of CTCs in the blood. Expansion of CTC numbers in cell culture systems has been a significant technical challenge. Cayrefourcq et al. recently described the first successful establishment of cell cultures and a permanent cell line from CTCs of a CRC patient, which has allowed for functional studies on the biology of CTCs as well as drug testing [9•].
CTC clusters are an even more rare entity in the peripheral blood of cancer patients, comprising only 2–5 % of detected CTCs. Aceto et al. recently demonstrated that breast and prostate cancer CTC clusters originate from oligoclonal groups of cells from the primary tumor and have a 23- to 50-fold increased metastatic potential compared to single CTCs [10•]. Unfortunately, most commercially available CTC detection methods, including CellSearch, do not provide specific results regarding the number of CTC clusters as part of standard clinical reports. A multitude of diverse technological platforms are being studied and developed to tap into the wealth of information from CTC clusters [11, 12].
In addition to CTCs, other cell types such as circulating cancer-associated macrophage-like cells (CAMLs) and tumor-educated blood platelets (TEPs) are being studied as candidate biomarkers of liquid biopsies [13, 14]. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) may be used as a means to target CTCs before their attachment and colonization at new sites [15]. Although early results are encouraging, CTC liquid biopsies are not yet widely used in daily clinical practice. Further investigation is necessary to establish the accuracy, reliability, and utility of various CTC-based testing. Once their robustness is validated in large, prospective studies of various clinical applications, CTC liquid biopsies will undoubtedly become a part of standard patient care in the future.
Technologies for Isolation and Characterization of CTC
CTCs are an ultra-rare event with a significant role in cancer. In most cases, there is less than one CTC in 1 ml of whole blood, which is on the scale 1 CTC in the background of 107 normal blood cells. DNA analysis, on the other hand, requires the ratio of tumor cell to normal cell to be at least 1–10 %. To overcome this challenge, multiple platforms have been developed for CTC enrichment and detection. These enrichment strategies are mainly based on the biological and/or physical properties of CTCs; detection can be achieved using immunological, molecular, or functional assays.
-
1.
CTC detection without enrichment
Some studies have analyzed all peripheral blood cells without CTC enrichment. Krivacic et al. described a method using fiber-optic array scanning (FAST) technology to detect CTCs with a sensitivity of 98 % and specificity of 1.5 × 10−5 [16]. Unlike enrichment approaches, there was no additional sample processing required for FAST cytometry that could result in reduced sensitivity due to cell loss. In 2015, Campton et al. described and evaluated the performance of a comprehensive and highly sensitivity platform, the AccuCyte–CyteFinder system, for collecting, identifying, and collecting CTCs [17]. Nucleated cells were separated from blood with a density-based cell separation apparatus, AccuCyte, and transferred to microscopic slides. After staining, the slides were imaged using a digital scanning microscope, CyteFinder. Recovery rates of CTCs from four cancer cells lines spiked into whole blood were between 90 and 91 %. CTC detection from blood samples of patients with various cancers was compared with the FDA-approved CellSearch system. Patient sample CTC counts matched or exceeded CellSearch CTC counts. Campton et al. also showed that a single-cell retrieval device, CytePicke, can harvest individual CTCs from slides after identification with CyteFinder for further genomic analysis [17].
The Epic Platform is another strategy for CTC detection without enrichment [18]. Werner et al. demonstrated its accuracy, linearity, and sensitivity for enumeration of all CTC concentrations tested. It has high specificity with zero CTCs detected in 18 healthy donor samples. In a clinical feasibility study, at least one traditional CTC per milliliter was detected in 89 % of 44 metastatic castrate-resistant prostate cancer samples. The Epic Platform was also able to detect additional CTC subpopulations, including CTC clusters, CK- CTCs, small CTCs, and apoptotic CTCs [18]. The main advantage of these CTC detection strategies without enrichment is the lack of bias associated with the selection process and elimination of possible CTC loss during blood cell depletion and other enrichment steps. A potential disadvantage is that there may not be enough viable CTCs recovered for further drug sensitivity testing or functional assays.
-
2.
Enrichment strategies based on CTC physical properties
Many CTC detection and isolation devices enrich CTCs on the basis of cell size, deformability, density, and electric charges. Different shapes of microfluidic devices have been developed to enrich CTCs with at least 80 % isolation efficiency [19]. The flexible micro spring array (FMSA) enriches CTCs based on a cell size and deformability difference between normal cells and cancer cells. FMSA can detect CTCs and CTC clusters in almost four times more patient samples as compared to CellSearch [20]. Enriched CTCs can be cultured or implanted into mice to form tumors in vivo [21]. Alternatively, the CellSieve™ microfilter enriches CTCs solely based on cell size [22]. The CellSieve microfilter can detect more CTCs as compared to CellSearch and detects CAMLs, which have a participatory role in tumor cell migration [13]. The isolation by size of epithelial tumor cells (ISET) method enriches CTCs by size-based filtration, and further CTC characterization is achieved with the laser microdissection technique [23]. The Parsortix system is a size and compressibility-based platform for CTC isolation. The purity of CTCs harvested by Parsortix at 3.1 % was significantly higher than IsoFlux at 1.0 %. Parsortix-isolated CTCs can be viable [24]. The Oncoquick system can enrich CTCs by density gradient centrifugation based on CTCs having a lower buoyant density than normal cells [25, 26]. CTCs can be enriched approximately 500 times, and the recovery rate is more than 90 % when 4 or 20 cells are spiked in blood [27]. The ApoStream™ device captures CTCs based on CTC biophysical characteristics using dielectrophoretic technology in a microfluidic flow chamber. The viability of isolated CTCs is greater than 97 %, and high-throughput is achieved [28]. The acoustic microfluid devices enrich and isolate CTCs by using sound waves to divert high-quality viable CTCs to a different flow channel with a recovery rate of approximately 83 % [29, 30]. The advantage of these strategies is that viable CTCs are collected with affordable simple devices. A disadvantage is biased collection using detection strategies based on known CTC physical properties resulting in CTC loss. Because these strategies are based on particular assumptions regarding CTC physical properties, they can potentially miss biologically relevant CTC subpopulations and hinder analysis of CTC heterogeneity.
-
3.
Enrichment strategies based on CTC biological properties
Most CTC enrichment and isolation platforms including the FDA-approved CellSearch method are based on biological properties of CTCs. Positive enrichment and negative enrichment are two strategies that are implemented. Positive enrichment uses CTC markers to capture CTCs and release normal cells while negative enrichment uses normal cell markers to capture normal cell and release CTCs. Epithelial cell adhesion molecule (EpCAM) immunoaffinity capture is frequently used as a positive selection method. The CellSearch system uses EpCAM-coated ferromagnetic beads to positively enrich CTCs [31]. Enriched CTCs can be transferred to the DEPArray system to isolate purified single cells for single-cell whole-genome amplification [32, 33]. The MagSweeper device enriches CTCs with EpCAM-coated magnetic beads using a magnetic rod. Isolated CTCs can be extracted individually for further analysis. Talasaz et al. showed that the the MagSweeper device was able to detect CTCs from all 47 tubes of blood from patients with metastatic breast cancer, but no CTCs were detected in any of the samples from five healthy donors [34]. Another method by AdnaGen uses immunomagnetic beads coated with a combination of antibodies to MUC1 and EpCAM to enrich CTCs and multiplex RT-PCR assays to detect CTCs. This method was shown to detect CTCs with high sensitivity and specificity with a detection limit of two tumor cells [35].
Although nucleic acid-based CTC detection has very high sensitivity, its specificity and accuracy are relatively low. Some positive selection platforms such as the Ephesia CTC-chip and IsoFlux combine immunomagnetic capture with microfluidic control to enhance CTC isolation [36, 37]. CTCs are detected and can be captured with an in vivo photoacoustic flow cytometry (PAFC) and functionalized plasmonic nanoparticles in a preclinical animal model [38]. The EpCAM-antibody-functionalized structured medical Seldinger guidewire (FSMW) method was utilized to capture CTCs in vivo. The FSMW captured no CTCs in healthy volunteers but 1–50 CTCs in 10 out of 12 breast cancer patients (83.3 %) and 2–515 CTCs in all of the 12 NSCLC patients [39].
Negative enrichment is most frequently achieved using CD45 as the negative selection marker. The RosetteSep™ CTC Enrichment Cocktail Containing Anti-CD36 enriches small cell carcinoma CTCs by negative selection with antibodies targeting CD2, CD16, CD19, CD36, CD38, CD45, CD66b, and glycophorin A. The RosetteSep™ CTC Enrichment Cocktail Containing Anti-CD56 enriches breast cancer CTCs by negative selection with antibodies targeting CD3, CD14, CD16, CD19, CD38, CD45, CD56, CD61, CD66b, and glycophorin A. Although the purity of enriched CTCs can be improved, they are viable and able to establish CTC cell lines [9•]. The geometrically activated surface interaction (GASI) chip also uses a negative selection strategy to enrich CTCs. Leukocytes are captured inside a channel coated with CD45 antibodies, and CTCs are released to the outlet. Nonlabeled, intact, and heterogeneous CTCs can be isolated regardless of EpCAM expression [40].
CTCs are highly heterogeneous including epithelial tumor cells, epithelial-to-mesenchymal cells, hybrid epithelial/EMT cells, and cancer stem cells. Using positive selection with EpCAM, high-purity CTCs are detected but selection bias may be induced. Reversibly, removing CTCs from the Immunoaffinity tag or surface can be challenging. Alternatively, using negative enrichment strategies can compromise CTC purity but heterogeneous CTCs or subpopulations of CTCs can potentially be harvested. It would be optimal to choose an enrichment strategy based upon the aim of further analysis. The new-generation CTC-iChip isolates CTCs using strategies that are either dependent or independent of EpCAM. Hence, it is applicable for any cancer whether it is of epithelial origin or not [41]. In addition to further direct analysis of CTCs, these unmanipulated CTCs can be cultured or used to establish patient-derived xenografts (PDXs) [42]. The two-stage microfluidic chip includes a microfluidic magnetic activated cell sorting (μ-MACS) chip and a GASI chip. The μ-MACS chip uses a negative selection strategy to deplete normal cells, and GASI classifies heterogeneous CTCs based on their characteristics [43]. Deneve et al. described the successfully isolation viable CTCs from the liver of CRC patients with the EpCAM independent CK19-Epispot (epithelial immunospot) assay which includes both negative selection with RosetteSep (CD45) and positive selection with CK19 [44]. These platforms are expected to facilitate further research in CTC heterogeneity.
Clinical Applications of CTCs in Colorectal Cancer
Given the minimally invasive nature of obtaining CTCs and the ability for serial monitoring through blood samples, research on CTCs remains a very active field. There is great interest in developing methods to extract and analyze CTCs as tools to aid in diagnosis and clinical management of CRC.
CTC enumeration has been established as a prognostic marker for CRC [6, 45–48]. In a large meta-analysis including 12 studies between 1998 and 2011, the presence of CTCs in patients with metastatic colorectal cancer (mCRC) correlated with shorter progression-free survival (PFS) and overall survival (OS) [45]. Groot-Koerkamp et al. found that mCRC patients with detectable CTCs had a 2.5-fold increased chance of death and a twofold increased chance of disease progression or recurrence [45]. A pivotal trial establishing the role of CTC enumeration as a prognostic and predictive marker was reported by Cohen et al. in 2008 [6]. This prospective multicenter study involved 430 patients with mCRC receiving first-, second-, and third-line systemic therapy and used the CellSearch® assay for CTC enumeration. Patients with baseline unfavorable CTC counts, defined as ≥3 CTCs/7.5 mL, compared to patients with favorable baseline counts (<3 CTCs/7.5 mL) had shorter median PFS (4.5 vs 7.9 months; P = 0.0002) and OS (9.4 vs 18.5 months; P < 0.0001). A small Japanese study had similar findings and demonstrated that ≥3 CTCs/7.5 mL during treatment was an independent predictor of PFS and OS in patients with mCRC [48]. In the non-mCRC setting, Bork et al. recently demonstrated the presence of CTCs (≥1 CTC/7.5 mL) in the preoperative setting is associated with worse OS compared to undetectable CTCs (38.4 vs 49.8 months; P < 0.001) [5•]. On multivariate analysis, preoperative CTC detection was a strong prognostic marker in non-mCRC [5•].
CTC enumeration has also been established as an indicator of treatment response [6, 48, 49•, 50, 51]. Conversion of high to low CTC counts for patients on therapy is associated with improved outcomes for CRC patients. Cohen et al. found that conversion of baseline unfavorable CTC counts to favorable at 3 to 5 weeks of therapy was associated with a significantly longer PFS (6.2 vs 1.6 months; P = 0.02) and OS (11.0 vs 3.7 months; P = 0.0002) as compared to patients with persistently unfavorable CTC counts [6]. Moreover, they reported a prognostic synergy between CTC enumeration and imaging response as defined by the Response Criteria in Solid Tumors (RECIST). Matsusaka et al. showed that patients with ≥3 CTCs at 2 and 8–12 weeks after initiation of oxaliplatin-based chemotherapy had shorter median PFS and OS than patients with <3 CTC counts [48]. Patients with CTC counts ≥3 at baseline with a decrease in the CTC count to <3 during treatment had a median PFS that was similar to patients with persistently low CTC counts. They concluded that a decrease in CTC count to <3 at 2 weeks after initiating chemotherapy was an indicator of treatment efficacy [48]. Alternatively, CTCs may be valuable in identifying patients with chemotherapy-resistant disease who have persistently elevated CTC counts during therapy [48]. Sastre et al. reported that patients with a CTC count <3 after three cycles of XELOX (capecitabine and oxaliplatin) with or without bevacizumab had a higher response rate than those with a CTC count ≥3 after three cycles (53 vs 26 %; P = 0.017) [49•]. Similarly, Kawahara et al. demonstrated that the conversion from detectable CTCs to undetectable with treatment correlated with decreased carcinoembryonic antigen (CEA) level and longer OS [50]. CTC enumeration may be used as an adjunct to standard methods for monitoring disease status, including CEA and imaging studies.
Based on these results, CTC enumeration may be a useful tool in stratifying patients and their response to therapy in future clinical trials. Multiple authors have proposed that patients could be divided based on changes in their CTC counts during treatment to represent different prognostic subgroups: patients without any detectable CTCs prior to and during treatment (longest PFS and OS), patients with high CTC counts at baseline which decreases during therapy (intermediate PFS and OS), and patients with persistently elevated CTC counts at baseline and during treatment (shortest PFS and OS) [49•, 50]. This stratification may one day help guide the clinical management of patients with mCRC.
The role of CTCs in early relapse detection is currently under investigation [52, 53]. Garrigós et al. reported a pilot study measuring CTCs in 16 patients with stages II and III surgically treated CRC during chemotherapy and follow-up [52]. Two of the 16 patients eventually developed relapsed disease and had elevated CTC counts, 5 and 6 months, respectively, prior to clinical evidence of relapse. One patient had an increase in CTC count without clinical evidence of relapse. A larger study with 90 stage III colon cancer patients evaluated the prognostic significance of CTCs detected after curative surgery and adjuvant chemotherapy (mFOLFOX) [53]. On multivariate analysis, Lu et al. found that persistently detectable CTC after therapy was an independent predictor of relapse and strongly correlated with reduced disease-free survival and OS [53]. These studies suggest that CTCs in early-stage CRC may play an important role in early detection of relapse, but further prospective studies are needed to validate these findings with standard methods for CTC detection and enumeration.
There is ongoing research on the utility of CTCs beyond enumeration, including analyzing molecular information or biomarkers to aid in clinical management. Cytokeratin-20 (CK20) and survivin expression in CTCs is currently being studied as a prognostic biomarker. Wong et al. demonstrated that the number of CK20-positive CTCs in CRC patients was associated with the stage of the disease and predicted metastasis and recurrence (P < 0.001) [54]. Moreover, patients with preoperative CK20-positive CTCs >11 had a shorter median OS compared to those with ≤11 CTCs (P < 0.0001). A recent study by Ning et al. demonstrated that patients with high CTC CK20 expression or survivin expression had significantly shorter median OS than patients with low expression of either marker [55•]. Patients who had detectable CTCs with high CK20 or survivin expression have inferior OS compared to those with low expression of both markers (HR, 4.39; 95 % CI 1.56–12.35; adjusted P = 0.005). This study shows that CTC CK20 and survivin expression may be effective biomarkers in predicting OS in mCRC patients receiving systemic therapy. Other potential prognostic biomarkers include CEA, cytokeratin 19 (CK19), and CD133. In a multicenter study with 735 patients, Iinuma et al. found that patients with CTCs expressing CEA, CK19, CK20, and CD133 mRNA had significantly worse DFS and OS than those with CTCs without these specific markers (P < 0.001) [56]. In particular, for patients with Duke’s B and C CRC, CTC expression of CEA/CK/CD133 was a significant prognostic factor [56].
Since CTCs can be collected through serial blood samples, it is an attractive method for obtaining longitudinal molecular and genetic analyses of the tumor and aids in targeted therapy investigations. It could be a minimally invasive approach to identify drug sensitivity or resistance-associated markers to guide therapeutic decisions. KRAS mutations in exon 2 (codons 12 and 13) are established as a negative predictive marker for treatment with EGFR inhibitors [57]. Several studies have successfully detected mutations in KRAS and PI3KCA from CTCs isolated from patients with CRC [58, 59•]. Heitzer et al. conducted the first comprehensive genetic profiling of CTCs using array-comparative genomic hybridization (CGH) and next-generation sequencing (NGS) [59•]. Deep sequencing analysis revealed that mutations in known driver genes (APC, KRAS, and PIK3CA) found in the primary tumor and metastasis were detected in corresponding CTCs. Moreover, other mutations that were initially found only in CTCs were also present at a subclonal level in the primary tumor and metastasis [59•]. With molecular characterization of CTCs, Gasch et al. demonstrated considerable intra-patient KRAS mutation heterogeneity with the same patient having both KRAS wild-type and KRAS-mutated CTCs [58]. Schölch et al. reported similar findings, specifically patients with KRAS wild-type primary tumors had both KRAS wild-type and KRAS mutant CTCs [60]. Upon deep sequencing of the primary tumor, mutations which were initially detected in CTCs were also found in the tumor. It is unclear at this time if this heterogeneity affects outcomes, but it is possible that failure of EGFR-inhibition in patients thought to have a KRAS wild-type CRC (determined from an arbitrary section of the primary tumor) may be partly due to the presence of a KRAS-mutated subclonal population. In a recent study, Buim et al. demonstrated that KRAS mutations derived from CTCs correlate with mutations in the primary tumor with a concordance of 71 % of matched cases (P = 0.017) [61]. These studies suggest that CTCs may play a role as a surrogate for primary tumors and metastasis when genomic analysis is necessary and the primary tumor is not available or as a means to serially monitor tumor genomes that can evolve during relapse, treatment, and progression. The ability to perform whole genome analysis from CTCs has the potential to uncover biomarkers predictive of sensitivity or resistance to available and investigational targeted agents, allowing for personalized therapies and appropriate patient selection to guide management of CRC. Thus, CTC enumeration and characterization may become an important companion diagnostics in drug development.
In the era of precision medicine, an in-depth investigation of CTCs is an attractive avenue for understanding the biology of a patient’s tumor. New technologies are being developed to allow for high-yield capture of CTCs and ex vivo expansion of isolated CTCs. The recently reported successful establishment of cell cultures and permanent cell line from CTCs derived from a colon cancer patient opens a myriad of opportunities [9•]. These cells can be characterized at the genome, transcriptome, proteome, and secretome levels, allowing for a variety of functional studies in the biology of CTCs as well as in vitro and in vivo drug testing.
Challenges remain in CTC research. New technologies are currently still being developed to optimize CTC isolation methods and require validation and standardization. Moreover, with the current available technologies, the use of CTC to monitor minimal residual disease in patients without overt evidence of metastasis remains challenging as CTC counts are typically very low in these patients. Although there are a variety of studies suggesting potential benefits from CTCs, their clinical utility remains unclear. There have not been randomized prospective interventional studies investigating how and what treatment changes can be made based on CTC enumeration and characterization in patients with CRC. Initiation of these studies may require validation and standardization of CTC isolation methods and analysis. Particularly challenging aspects include robust technologies to evaluate genomic sequences at a single cell level and the ability to culture isolated CTCs for further studies.
Circulating Tumor DNA
Cancer development and treatment is a dynamic process. Primary tumors may not reflect the current or complete tumor molecular profile. Molecular readouts are more specific indicators of the disease process. ctDNA refers to small DNA fragments that are shed by the tumor into the bloodstream. It is defined by mutations and other genomic changes that are hallmarks of cancer cell and is a potential surrogate for the entire tumor genome. CTCs reflect more of the metastases-initiating cells while ctDNA represents more of the tumor burden. Recent interest in liquid biopsies has shifted to the study of ctDNA. ctDNA outperforms CTCs for KRAS mutation detection in both diagnostic sensitivity and specificity. A ctDNA test can detect progression of breast cancer 5 months before radiographic evidence, which would potentially allow an ineffective therapy to be abandoned earlier [62]. When comparing CTCs and ctDNA in the detection of KRAS mutation in patients undergoing surgery for suspected lung cancer, ctDNA testing had a greater diagnostic sensitivity and specificity than testing for DNA extracted from CTCs, suggesting that ctDNA may be the preferential specimen type for mutation screening [63]. Comparing ctDNA (97 %) with CTC (87 %) and protein marker CA 15-3 (78 %) detection rates from patients in whom somatic genomic alterations were identified, ctDNA levels showed a greater detection rate and greater correlation with changes in tumor burden; it also provided the earliest measure of treatment response in 53 % subjects. ctDNA is an informative, inherently specific, and highly sensitive biomarker for human cancer [62]. It remains in the circulation for a few hours before being metabolized, which allows real-time monitoring of tumors as they spread and mutate or develop resistance to treatment [64]. ctDNA carries the evolutionary information of both primary and metastatic tumors when resistant CRC cells dynamically evolve in response to intermittent drug treatment [65•]. Comparing biopsy and plasma samples from the same patient shows that ctDNA can allow real-time sampling of multifocal clonal evolution [66]. Moreover, ctDNA analysis can be used to track tumor burden and analyze cancer genomes and metastatic heterogeneity non-invasively.
Technologies for Detection and Analysis of ctDNA
The techniques and technology for detection, quantification, and analysis of ctDNA have vastly improved over recent years. A variety of methods have been used for purification of ctDNA including modified salting-out, chromatography resins, magnetic beads, and guanidium thiocyanate. However, more recently, this process has been simplified with the availability of various commercial kits, such as QIAmp 96 spin blood DNA extraction kit, QIAmp DNA blood mini kit, Bilatest DNA kit, Quanti-iT™ DNA high-sensitivity assay kit. Sonnenberg et al. recently described a novel electrokinetic technique with an AC electrokinetic microarray device (Biological Dynamics) that allows for rapid isolation of circulating DNA from blood [67]. Unfortunately, these isolation and detection procedures are not standardized, and none of these technologies are FDA approved. Moreover, ctDNA is present at very low concentrations in peripheral blood and not easily enriched. Levels of ctDNA vary widely from person to person and can be hard to detect in early stages of cancer. Recently, a ctDNA enrichment process was developed as the first non-invasive method that allowed for high-yield isolation directly from a small volume of unprocessed plasma with significant increase in isolation efficiency and more than 20-fold recovery [68]. Because of its successful enrichment of ctDNA, this strategy described by Yeh et al. allowed for accurate NGS genomic analyses directly from droplet volumes of plasma [68].
There are two general approaches to ctDNA analysis. The first is a targeted approach that involves identification of specific mutations in the primary tumor and analysis of these known genetic changes as circulating DNA. The alternative untargeted approach is to scan regions of DNA extracted from plasma or serum for mutations of interest in a blinded manner without knowledge of genetic changes in the primary tumor.
With recent technological advancements, a myriad of highly sensitive techniques are available for the detection and analysis of mutant alleles present in the circulation at very low frequencies. These targeted methods include amplification refractory mutation system (ARMS) [69]; digital polymerase chain reaction (dPCR) [70, 71•, 72]; beads, emulsions, amplification, and magnetics (BEAMing) [73, 74]; and pyrophosphorolysis-activated polymerization (PAP) [75]. Bettegowda et al. reported using digital methods, including dPCR and the Safe-Sequencing System (Safe-SeqS) to evaluate the ability of ctDNA to detect tumors in 640 patients with various cancer types. In a subgroup of patients with mCRC, the sensitivity of ctDNA for detecting clinically relevant KRAS mutation using the aforementioned methods was 87.2 % with a specificity of 99.2 % [76].
Another highly specific approach to detecting low-frequency mutations in the circulation was described by Forshew et al. in 2012. Using tagged-amplicon deep sequencing (TAm-Seq), they identified cancer-related mutations present in circulating DNA at allele frequencies as low as 2 % with sensitivity and specificity of more than 97 % [77•].
In 2014, Newman et al. reported on the use of cancer personalized profiling by deep sequencing (CAPP-Seq), a cost-effective and ultrasensitive method for quantification of ctDNA, in the diagnosis and management of non-small cell lung cancer (NSCLC) [78•]. CAPP-Seq combines optimized library preparation methods for low DNA input masses with a multiphase bioinformatics approach to design a selector consisting of biotinylated DNA oligonucleotides that target mutated regions in a specific cancer type. The selector is initially applied to tumor DNA to identify a patient’s specific genetic aberrations and then to circulating DNA to quantify ctDNA [78•]. Using this method, they detected ctDNA in 100 % of stage II–IV NSCLC with 96 % specificity for mutant allele fractions down to about 0.02 % [78•].
Kidess et al. developed an assay based on sequence-specific synchronous coefficient of drag alteration (SCODA) technology, which enables efficient enrichment of ctDNA to detect specific mutations with high sensitivity and specificity [79]. They analyzed tissue and plasma samples of patients with non-metastatic and mCRC and focused on 46 mutations in 4 genes (BRAF, EGFR, KRAS, and PIK3CA). The assay demonstrated a limit of detection of 0.001 % for most of the 46 mutations. Detected mutations were concordant in tissue and plasma for 93 % of mCRC patients. An expanded assay with a larger panel of mutations is currently under development.
The untargeted approach to ctDNA analysis includes whole genome sequencing and exome sequencing. Whole genome sequencing can be done using next-generation sequencing-based approaches. Leary et al. described a whole genome sequencing-based method called personalized analysis of rearranged ends (PARE) to identify tumor-derived structural alterations through analysis of ctDNA from colorectal and breast cancer patients [80]. Detected alterations using massively parallel sequencing included chromosomal copy number changes and rearrangements, such as amplification of cancer driver genes ERBB2 and CDK6. Since most cancers have multiple chromosomal abnormalities unlikely to be present in normal cells, this approach is highly specific, although the sensitivity largely depends on the amount of sequence data obtained [80]. In 2013, Murtaza et al. described whole exome sequencing of ctDNA in plasma samples to track genetic evolution of metastatic cancer in response to therapy [81•]. Quantification of allele fractions identified increased representation of mutation alleles associated with emergence of therapy resistance. This proof of principle study showed that exome analysis of plasma ctDNA is feasible and allows for non-invasive characterization of tumor evolution. These untargeted approaches to ctDNA analysis allow for broad patient coverage because these methods do not rely on recurrent genetic changes. However, disadvantages include lack of analytical sensitivity and detection limitations.
Clinical Applications of ctDNA in Colorectal Cancer
Similar to CTCs, ctDNA is currently being investigated as a minimally invasive cancer biomarker. Released as fragments from necrotic and apoptotic tumor cells, ctDNA carries tumor-related genetic and epigenetic alterations that provide information on relapse, treatment response, drug sensitivity or resistance, and prognosis. ctDNA can serve as an additional tool in the management of patients with CRC.
Given the low levels of CTCs in most CRC patients, especially in the non-metastatic setting, detection of CTCs requires more cumbersome isolation and enrichment techniques; ctDNA, on the other hand, is more easily detected in plasma or serum. In 2005, Diehl et al. showed that detection and quantification of circulating APC gene fragments is feasible and can serve as a potential biomarker for CRC [82]. Circulating APC gene fragments (wild type and mutant) were elevated in patients with CRC, especially in patients with advanced CRC. However, there was no correlation between tumor burden and the concentration of APC fragments or the percentage of circulating mutant APC fragments [82].
Studies have shown that ctDNA can serve as a potential molecular marker for poor clinical outcomes in CRC patients and as a tool for early detection of recurrence or metastatic disease [82–84]. Wang et al. reported that detection of APC, KRAS, and p53 mutations in DNA extracted from the serum of patients with CRC correlated with significantly higher postoperative metastasis/recurrence rate than in patients without detectable ctDNA (P < 0.001) [83]. In 2006, Frattini et al. reported that quantification of ctDNA may be useful for monitoring early stage CRC patients postoperatively for detection of relapse [84]. They found that ctDNA levels were significantly higher in patients with CRC compared to healthy controls. In tumor-free CRC patients, ctDNA levels decreased progressively during the follow-up period, while patients with recurrence or metastasis had increasing levels [84]. Another study with 18 patients undergoing multimodality therapy for CRC demonstrated that ctDNA quantification can be used to monitor tumor dynamics in patients undergoing surgery or chemotherapy [73]. All but one patient with detectable ctDNA in the postoperative period (13–56 days after surgery) had evidence of relapse within 1 year, whereas patients with undetectable ctDNA had no recurrence (P = 0.006). Diehl et al. also compared ctDNA to CEA levels as a biomarker for disease and reported that detectable ctDNA is more sensitive than CEA (ctDNA 100 % vs CEA 56 %; P = 0.008). Detection of ctDNA is a better predictive marker of disease recurrence than CEA in the postoperative setting (P = 0.003). In the advanced CRC setting, Schwarzenbach et al. showed that these patients had high levels of ctDNA that fluctuated during chemotherapy and high ctDNA levels significantly correlated with a shorter OS (P = 0.02) [85].
Qualitative alterations in ctDNA, including molecular mutations, integrity, and methylation, can provide information on drug sensitivity, drug resistance, prognosis, and treatment response [65•, 77•, 81•, 86•, 87•, 88]. Using targeted deep DNA sequencing analysis of ctDNA, mutations in certain oncogenes and tumor suppressors can be identified. ctDNA with KRAS mutations have been identified in patients with initial KRAS wild-type primary tumors after treatment with EGFR inhibitors, cetuximab, and panitumumab, indicating that failure of EGFR inhibition may be due to the evolution of KRAS mutations in select patients [86•, 87•]. Diaz et al. demonstrated that 9 out of 24 (38 %) patients with KRAS wild-type primary tumors receiving single-agent panitumumab developed detectable KRAS-mutated ctDNA about 5–6 months after initiation of therapy. Three patients developed multiple different KRAS mutations [86•]. Misale et al. showed that the emergence of secondary drug resistance to EGFR inhibitor, cetuximab, was likely due to the emergence of mutant KRAS ctDNA. KRAS mutant alleles were detected in the circulation as early as 10 months before radiographic evidence of disease progression. The use of ctDNA to monitor for the evolution of KRAS mutant ctDNA may allow for early identification of individuals at high risk for developing drug resistance to anti-EGFR therapy prior to radiographic evidence of disease progression [87•]. In 2015, Siravegna et al. used ctDNA to track clonal evolution during treatment with EGFR inhibitors and identified alterations in various genes, including KRAS, NRAS, MET, ERBB2, FLT3, EGFR, and MAP2K1 [65•]. Interestingly, they found that the concentration of mutated KRAS ctDNA clones, which emerged during anti-EGFR therapy, declined upon withdrawal of EGFR inhibitors. Pharmacogenomic analysis of CRC cells that had acquired resistance to cetuximab demonstrated that the withdrawal of anti-EGFR antibodies resulted in a decline in KRAS-mutated clones and the population regained drug sensitivity [65•]. These findings suggest that the genome of CRC cells adapts dynamically to the targeted therapy and provides an explanation for the efficacy of re-challenge with EGFR blockade. Moreover, it shows that ctDNA can be used to monitor tumor dynamics and guide management.
Although there is active research in the potential roles of ctDNA, there is currently no validated platform or standardized method for ctDNA extraction and analysis. Its utility in the clinical setting remains controversial. There may be elevations of ctDNA which reflect physiologic or pathologic processes that are not disease-specific. Fleischhacker et al. showed that increased levels of ctDNA may be found in patients with benign lesions, inflammatory disease, or tissue trauma [89]. In addition, the analysis of ctDNA is limited. While analysis of CTCs can be done at the genome, transcriptome, and proteasome level with functional in vivo and in vitro assays, ctDNA can only be analyzed at the genomic level with molecular DNA assays, including NGS.
Conclusions and Future Outlook
As liquid biopsies, CTC and ctDNA capture the heterogeneity across tumor sites and the evolution of tumor cells and mutations. CTC and ctDNA reflect biologically different aspects of disease. For early detection of cancer and to identify the origin of carcinoma of unknown primary, CTC holds promise. CTC enumeration alone is prognostic and monitors responses to therapy in CRC, breast cancer, and prostate cancer [90]. CTCs can provide evaluable tumor cells for phenotyping, genotyping, primary cell line culture, and patient-derived xenograft models [91]. With the addition of cell culture supplements that cover critical signaling pathways to support CTC survival and proliferation, in vitro culture of CTC and organoid culture can be tested for drug sensitivity to guide treatment directly [42, 92]. With improved CTC acoustic separation and microfluidic isolation technologies, genomic analysis of CTCs and even single cell genomics are now feasible. On the other hand, ctDNA provides real-time molecular information to monitor treatment response and relapse. It holds great promise to unravel drug-resistance mechanisms. Multiple mutation analysis of ctDNA via NGS could become a gold standard of modern cancer management. These two liquid biopsy biomarkers contribute complementary information, and therefore, utilitizing both approaches to study tumor metastasis and treatment resistance is warranted. Precision medicine is changing clinical practice by tailoring treatment based on an individual’s genetic makeup. CTC and ctDNA will complement biopsies as diagnostic procedures and hopefully will make cancer treatment more precise.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
American Cancer Society. Cancer facts & figures 2015. Atlanta: American Cancer Society; 2015.
American Cancer Society. Colorectal cancer facts & figures 2014-2016. Atlanta: American Cancer Society; 2014.
Ashworth TR. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J. 1869; 146-149.
Mandel P, Metais P. Les acides nucleiques du plasma sanguin ches l’homme. C R Seances Soc Biol Fil. 1948;142(3-4):241–3.
Bork U et al. Circulating tumour cells and outcome in non-metastatic colorectal cancer: a prospective study. Br J Cancer. 2015;112(8):1306–13. Prospective study evaluating the role of CTCs in patients with non-metastatic CRC. Preoperative CTC detection is a strong prognostic marker and is associated with worse OS in the non-metastatic CRC setting.
Cohen SJ et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213–21.
Cristofanilli M et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–91.
de Bono JS et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302–9.
Cayrefourcq L et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015;75(5):892–901. Study describing the first successful establishment of cell cultures and a permanent cell line from CTCs of a CRC patient. The cells have been characterized at the genome, transcriptome, proteome, and secretome levels.
Aceto N et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. Study to define the origin and functional properties of CTC clusters. CTC clusters originate as oligoclonal groups of cells from the primary tumor that exhibit increased metastatic propensity compared to single CTCs. Abundance of CTC clusters in the peripheral blood is associated with adverse outcomes.
Cho EH et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol. 2012;9(1):016001.
Stott SL et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A. 2010;107(43):18392–7.
Adams DL et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc Natl Acad Sci U S A. 2014;111(9):3514–9.
Best MG et al. RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28(5):666–76.
Goldberg GS et al. Global effects of anchorage on gene expression during mammary carcinoma cell growth reveal role of tumor necrosis factor-related apoptosis-inducing ligand in anoikis. Cancer Res. 2001;61(4):1334–7.
Krivacic RT et al. A rare-cell detector for cancer. Proc Natl Acad Sci U S A. 2004;101(29):10501–4.
Campton DE, Ramirez AB, Nordberg JJ, Drovetto N, Clein AC, Varshavskaya P, et al. High-recovery visual identification and single-cell retrieval of circulating tumor cells for genomic analysis using a dual-technology platform integrated with automated immunofluorescence staining. BMC Cancer. 2015;15:360.
Werner SL et al. Analytical validation and capabilities of the Epic CTC platform: enrichment-free circulating tumour cell detection and characterization. J Circ Biomark. 2015;4:3.
Tan SJ et al. Microdevice for the isolation and enumeration of cancer cells from blood. Biomed Microdevices. 2009;11(4):883–92.
Harouaka RA et al. Flexible micro spring array device for high-throughput enrichment of viable circulating tumor cells. Clin Chem. 2014;60(2):323–33.
Gallant JN et al. Predicting therapy response in live tumor cells isolated with the flexible micro spring array device. Cell Cycle. 2013;12(13):2132–43.
Adams DL et al. Cytometric characterization of circulating tumor cells captured by microfiltration and their correlation to the CellSearch® CTC test. Cytometry A. 2015;87(2):137–44.
Pinzani P et al. Isolation by size of epithelial tumor cells in peripheral blood of patients with breast cancer: correlation with real-time reverse transcriptase-polymerase chain reaction results and feasibility of molecular analysis by laser microdissection. Hum Pathol. 2006;37(6):711–8.
Xu L et al. Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS One. 2015;10(9), e0138032.
Kaifi JT et al. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: a prospective trial with different detection techniques. Cancer Biol Ther. 2015;16(5):699–708.
Obermayr E et al. Assessment of a six gene panel for the molecular detection of circulating tumor cells in the blood of female cancer patients. BMC Cancer. 2010;10:666.
Clawson GA et al. Circulating tumor cells in melanoma patients. PLoS One. 2012;7(7), e41052.
Gupta V et al. ApoStream™, a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics. 2012;6(2):24133.
Antfolk M et al. Acoustofluidic, label-free separation and simultaneous concentration of rare tumor cells from white blood cells. Anal Chem. 2015;87(18):9322–8.
Li P et al. Acoustic separation of circulating tumor cells. Proc Natl Acad Sci U S A. 2015;112(16):4970–5.
Miller MC, Doyle GV, Terstappen LW. Significance of circulating tumor cells detected by the cell search system in patients with metastatic breast colorectal and prostate cancer. J Oncol. 2010;2010:617421.
Swennenhuis JF, Terstappen L. Sample preparation methods following cell search approach compatible of single-cell whole-genome amplification: an overview. Methods Mol Biol. 2015;1347:57–67.
Pestrin M et al. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol. 2015;9(4):749–57.
Talasaz AH et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci U S A. 2009;106(10):3970–5.
Zieglschmid V et al. Combination of immunomagnetic enrichment with multiplex RT-PCR analysis for the detection of disseminated tumor cells. Anticancer Res. 2005;25(3A):1803–10.
Saliba AE et al. Microfluidic sorting and multimodal typing of cancer cells in self-assembled magnetic arrays. Proc Natl Acad Sci U S A. 2010;107(33):14524–9.
Harb W, et al. Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl Oncol. 2013; 6(5): 528–38.
Galanzha EI, Zharov VP. Circulating tumor cell detection and capture by photoacoustic flow cytometry in vivo and ex vivo. Cancers (Basel). 2013;5(4):1691–738.
Saucedo-Zeni N et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int J Oncol. 2012;41(4):1241–50.
Hyun KA, Lee TY, Jung HI. Negative enrichment of circulating tumor cells using a geometrically activated surface interaction chip. Anal Chem. 2013;85(9):4439–45.
Ozkumur E et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med. 2013;5(179):179ra47.
Yu M et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345(6193):216–20.
Hyun KA et al. Two-stage microfluidic chip for selective isolation of circulating tumor cells (CTCs). Biosens Bioelectron. 2015;67:86–92.
Deneve E et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem. 2013;59(9):1384–92.
Groot Koerkamp B et al. Circulating tumor cells and prognosis of patients with resectable colorectal liver metastases or widespread metastatic colorectal cancer: a meta-analysis. Ann Surg Oncol. 2013;20(7):2156–65.
Rahbari NN et al. Meta-analysis shows that detection of circulating tumor cells indicates poor prognosis in patients with colorectal cancer. Gastroenterology. 2010;138(5):1714–26.
Cohen SJ et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol. 2009;20(7):1223–9.
Matsusaka S et al. Circulating tumor cells as a surrogate marker for determining response to chemotherapy in Japanese patients with metastatic colorectal cancer. Cancer Sci. 2011;102(6):1188–92.
Sastre J et al. Circulating tumor cell count is a prognostic factor in metastatic colorectal cancer patients receiving first-line chemotherapy plus bevacizumab: a Spanish Cooperative Group for the Treatment of Digestive Tumors study. Oncologist. 2012;17(7):947–55. Prospective ancillary study of the Maintenance in Colorectal Cancer trial to evaluate the CTC count as a prognostics and/or predictive marker for efficacy endpoints. CTC enumeration is a strong prognostic factor for PFS and OS outcomes and an indicator of treatment response in mCRC patients receiving XELOX +/− bevacizumab.
Kawahara H et al. Determination of circulating tumor cells for prediction of recurrent colorectal cancer progression. Hepatogastroenterology. 2012;59(119):2115–8.
Tol J et al. Circulating tumour cells early predict progression-free and overall survival in advanced colorectal cancer patients treated with chemotherapy and targeted agents. Ann Oncol. 2010;21(5):1006–12.
Garrigós N et al. Circulating tumour cell analysis as an early marker for relapse in stage II and III colorectal cancer patients: a pilot study. Clin Transl Oncol. 2010;12(2):142–7.
Lu CY et al. Circulating tumor cells as a surrogate marker for determining clinical outcome to mFOLFOX chemotherapy in patients with stage III colon cancer. Br J Cancer. 2013;108(4):791–7.
Wong SC et al. Clinical significance of cytokeratin 20-positive circulating tumor cells detected by a refined immunomagnetic enrichment assay in colorectal cancer patients. Clin Cancer Res. 2009;15(3):1005–12.
Ning Y et al. Cytokeratin-20 and survivin-expressing circulating tumor cells predict survival in metastatic colorectal cancer patients by a combined immunomagnetic qRT-PCR approach. Mol Cancer Ther. 2015;14(10):2401–8. Study evaluating the prognostic value of baseline CTC CK20 and survivin expression in mCRC patients. CTC CK20 and survivin expression effectively predict OS in mCRC patients receiving chemotherapy.
Iinuma H et al. Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes’ stage B and C colorectal cancer. J Clin Oncol. 2011;29(12):1547–55.
Walther A et al. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9(7):489–99.
Gasch C et al. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem. 2013;59(1):252–60.
Heitzer E et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73(10):2965–75. This study conducted the first comprehensive genomic profiling of CTCs using CGH and next-generation sequencing. Genomic profiling of CTCs is feasible. Most mutations thought initially to be exclusive to CTCs were present at a subclonal level in the primary tumor and/or metastasis suggesting that CTCs can be used to elucidate changes in the tumor genome that was either not present or observed at the time of initial diagnosis and can be used to tailor management.
Schölch S et al. Circulating tumor cells of colorectal cancer. Cancer Cell Microenviron. 2014;1(5):1–6.
Buim ME et al. Detection of KRAS mutations in circulating tumor cells from patients with metastatic colorectal cancer. Cancer Biol Ther. 2015;16(9):1289–95.
Dawson SJ et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–209.
Freidin MB et al. Circulating tumor DNA outperforms circulating tumor cells for KRAS mutation detection in thoracic malignancies. Clin Chem. 2015;61(10):1299–304.
Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13(11):795–806.
Siravegna G et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):795–801. Study using ctDNA to genotype colorectal cancer tumors and track clonal evolution during treatment with EGFR-specific antibodies. This study showed that mutated KRAS clones that emerge in the blood during EGFR blockade decline upon withdrawal of EGFR-specific anitbodies, indicating clonal evolution and that the CRC genome adapts dynamically to intermittent drug schedules.
Murtaza M et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760.
Sonnenberg A et al. Rapid electrokinetic isolation of cancer-related circulating cell-free DNA directly from blood. Clin Chem. 2014;60(3):500–9.
Yeh CH et al. Next-generation sequencing analysis of high-quality and high-quantity cell-free circulating DNA prepared from droplet volumes of patient plasma. J Clin Oncol. 2015;33(15_suppl):abstr e22008.
Spindler KL et al. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18(4):1177–85.
Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–41.
Taly V et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59(12):1722–31. Study demonstrating the feasibility and utility of multiplex dPCR of plasma DNA to screen for 7 common mutations in KRAS codons 12 and 13 in patients with mCRC.
Sefrioui D et al. Clinical value of chip-based digital-PCR platform for the detection of circulating DNA in metastatic colorectal cancer. Dig Liver Dis. 2015;47(10):884–90.
Diehl F et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
Dressman D et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100(15):8817–22.
Liu Q, Sommer SS. Pyrophosphorolysis-activated polymerization (PAP): application to allele-specific amplification. Biotechniques. 2000;29(5):1072–6. 1078, 1080 passim.
Bettegowda C et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Forshew T et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68. Study evaluating TAm-Seq of ctDNA as a noninvasive method to screen for low-frequency mutations. This method identified cancer mutations present in ctDNA at allele frequencies as low as 2% with sensitivity and specificity >97%.
Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54. Study evaluating the utility and feasibility of CAPP-Seq to detect and quantify ctDNA with broad patient coverage. CAPP-Seq allows for highly sensitive and noninvasive detection of ctDNA in a majority of patients with NSCLC at low cost.
Kidess E et al. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget. 2015;6(4):2549–61.
Leary RJ et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4(162):162ra154.
Murtaza M et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12. Study evaluating exome-wide sequencing using ctDNA. Exome-wide sequencing of ctDNA is feasible and can be used to track genomic evolution of metastatic cancers in response to therapy.
Diehl F et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102(45):16368–73.
Wang JY et al. Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J Surg. 2004;28(7):721–6.
Frattini M et al. Quantitative analysis of plasma DNA in colorectal cancer patients: a novel prognostic tool. Ann N Y Acad Sci. 2006;1075:185–90.
Schwarzenbach H et al. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann N Y Acad Sci. 2008;1137:190–6.
Diaz LA et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–40. Study evaluating the evolution of mutant KRAS ctDNA in patients receiving panitumumab. Thirty-eight percent of patients whose tumors were initially KRAS wild type developed KRAS mutations detected by ctDNA. Mathematical modelling indicated that the mutations were present in expanded subclones before the initiation of panitumumab suggesting that the emergence of KRAS mutations is a mediator of acquired resistance to EGFR blockade and it is feasible to detect these mutations in a non-invasive manner.
Misale S et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6. Study using ctDNA to show that molecular alterations of KRAS are causally associated with the onset of acquired resistance to anti-EGFR treatment in CRC. KRAS mutant alleles were detectable in blood as early as 10 months before radiographic evidence of disease progression. The study identifies KRAS mutations as frequent drivers of acquired resistance to cetuximab in CRC and show that the emergence of KRAS mutant clones can be detected as ctDNA non-invasively months prior to radiographic progression.
Trevisiol C et al. Prognostic value of circulating KRAS2 gene mutations in colorectal cancer with distant metastases. Int J Biol Markers. 2006;21(4):223–8.
Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer—a survey. Biochim Biophys Acta. 2007;1775(1):181–232.
Das A et al. Clinico-pathological correlation of serial measurement of circulating tumor cells in 24 metastatic colorectal cancer patients receiving chemotherapy reveals interpatient heterogeneity correlated with CEA levels but independent of KRAS and BRAF mutation. Cancer Biol Ther. 2015;16(5):709–13.
Bertotti A et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–7.
Gao D et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–87.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Translational Colorectal Oncology
Rights and permissions
About this article

Cite this article
Tan, C.R.C., Zhou, L. & El-Deiry, W.S. Circulating Tumor Cells Versus Circulating Tumor DNA in Colorectal Cancer: Pros and Cons. Curr Colorectal Cancer Rep 12, 151–161 (2016). https://doi.org/10.1007/s11888-016-0320-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-016-0320-y