
Abstract
Fluoropyrimidine-based chemotherapy improves survival in stage III colon cancer patients in the adjuvant setting, whereas its clinical benefit in stage II is limited. Adjuvant therapy could be considered in patients with high-risk stage II disease, who are more likely to benefit from chemotherapy. Clinicopathological factors have been routinely used for risk stratification in stage II, as well as microsatellite instability (MSI) analysis, which has been recently incorporated in clinical guidelines as a prognostic marker. Other molecular markers, such as KRAS and BRAF mutations, suggested improving accuracy in prognostic classification in non-metastatic disease. Recent data derived from randomized clinical trial demonstrated that KRAS/BRAF gene mutations are associated with worse outcome depending on MSI status and tumor localization. Similarly, supervised gene expression signatures have refined recurrence risk stratification in several prospective studies although the clinical utility is still debatable. In this review, we focus on the new data on molecular and gene expression profiling in non metastatic colon cancer and the impact on prognostication and treatment decision.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most common cancer and the fourth-leading cause of cancer death worldwide [1]. Surgery is the standard of care in localized disease, and fluoropyrimidine-based adjuvant chemotherapy has clearly demonstrated survival benefit in stage III [2, 3], while survival improvement in stage II due to adjuvant therapy is limited [4]. Overall, adjuvant chemotherapy for patients with stage II colon cancer (CC) could be considered for those classified as high risk (including patients with inadequately sampled nodes and T4 lesions) [4, 5]. In an attempt to refine risk stratification, several molecular factors have been investigated as prognostic markers (like TP53 mutations, DNA ploidy, loss of heterozygosity on chromosome 18q, or thymidylate synthase expression). However, most of them could not be included in the decision-making process regarding the use of adjuvant chemotherapy because of inconclusive data, except for microsatellite instability [4, 6]. Apart from molecular factors, gene expression signatures developed as prognostic tools have been recently validated in the adjuvant setting [7–9]; thus, we should consider integrating them into treatment decision algorithm.
As a result of new informative data on molecular markers and gene expression assays, we aim to discuss their potential impact on clinical practice.
Molecular Factors
Identifying patients who could be managed without adjuvant chemotherapy is a matter of concern for the oncology community. The increasing knowledge on colorectal tumorigenesis has suggested several genomic alterations as putative prognostic factors but contradictory results have arised, mostly derived from retrospective studies [6]. Results from a retrospective study evaluating guanyl-cyclase C expression in lymph nodes in stage II CC have been reported recently suggesting that high levels of guanyl-cyclase C increased the risk of relapse [10]. Although this is an encouraging data, its prognostic value warrants further investigation. Microsatellite instability (MSI) has been widely investigated in cohorts of patients associated to prospective clinical trials with consistent results. There is currently sufficient evidence supporting that MSI is a marker of favorable outcome in stage II CC and consequently, mismatch repair or MSI testing either by immunohistochemistry or PCR is recommended by clinical guidelines [11, 12].
The mutational status of KRAS and BRAF has also been analyzed in large prospective clinical trials in the adjuvant setting, although their prognostic value is controversial. However, recent data derived from adjuvant clinical trials have prompted questions about their impact on outcome.
KRAS
The oncogene KRAS has been clearly associated to lack of efficacy to anti-EGFR drugs in several clinical trials in metastatic setting [13, 14], along with the presence of mutations in NRAS recently described [15, 16]. However, the prognostic value of KRAS mutations is not well established since the results published were not conclusive, either in the adjuvant or metastatic settings [6].
Recent studies provide new information on KRAS mutations and its association to survival in early disease. KRAS exon 2 mutations have been related to worse prognosis in stage III CC patients enrolled into the PETACC 8 and the NCCTG N0147 clinical trials [17–20]. Interestingly, KRAS mutations had a negative impact on disease-free survival (DFS) in patients with proficient DNA mismatch repair (pMMR) tumors [18]. Moreover, in the pooled analysis of microsatellite stable (MSS) stage III CC patients treated with oxaliplatin +/− cetuximab, KRAS mutations were also independent predictors of poor survival [20].
BRAF
BRAF is a proto-oncogene involved in the RAS-mitogen-activated protein kinase pathway and is mutated in 10 % of colorectal tumors, approximately. Regarding the role of BRAF mutations in CRC, either the negative predictive value to anti-EGFR therapies or the association to worse outcome in non-metastatic disease remains unclear [21–24]. Although it is recommended not to treat BRAF mutant mCRC patients with cetuximab or panitumumab [25, 26], several studies which might elucidate the predictive value of BRAF mutations are ongoing (ClinicalTrials.gov Identifier: NCT01704703; ClinicalTrials.gov Identifier: NCT01640444).
The prognostic value of BRAF mutations in locally advanced disease has also been analyzed in cohorts of patients from the NCCTG N0147 and the PETACC 8 clinical trials [18, 20, 27]. Patients with BRAF mutant tumors have significantly worse DFS than those patients with BRAF wild-type tumors [18]. However, this negative impact was only retained in the subgroup of patients with microsatellite stable tumors, suggesting that the favorable impact of defective MMR (dMMR) may be stronger than BRAF mutations.
Additionally, a combined molecular analysis was performed in order to better characterize high-risk stage III CC patients [27]. Five different subgroups were defined according to MMR, MLH1 hypermethylation, BRAF, and KRAS status. Forty-nine percent of tumors were pMMR and KRAS/BRAF wild type, 35 % were pMMR and KRAS mutant/BRAF wild type, 7 % were pMMR and KRAS wild type/BRAF mutant, 7 % were dMMR and BRAF mutant (or MLH1 hypermethylated), and 3 % were dMMR and BRAF wild type. Molecular subgroups were clinically differentiated as well as statistically significant differences were observed among them regarding outcome. Mutations in KRAS or BRAF in pMMR tumors significantly decreased DFS compared to wild-type KRAS/BRAF and pMMR tumors with a HR of 1.48 and 1.43, respectively. In conclusion, it was suggested that KRAS and BRAF mutations are poor prognostic markers in pMMR tumors, although this was not retained in BRAF mutant tumors located in distal colon (HR = 1.104, p value = 0.7318).
In summary, molecular studies on primary tumors from stage III CC patients enrolled into clinical trials have provided new insights in this setting. Despite this encouraging data, there is insufficient evidence to standardize KRAS and BRAF mutation analysis in locally advanced CC for treatment decision. However, we should consider including these markers as stratification factors in future clinical trials.
Gene Expression Signatures
Several gene expression profiles have been developed using gene expression data from different microarray platforms, and independent validation tests have further been performed in distinct CRC cohorts [28••, 29]. OncotypeDX® is the most widely validated one but other classifiers need further investigation. Veridex developed and validated a 23-gene signature in a microarray oligonucleotide platform with fresh tumor tissue from stage II CC patients [30]. The signature was eventually reduced to seven genes, translated onto a RT-PCR platform with paraffin-embedded (FFPE) tumor tissue and validated independently with fascinating results in terms of metastasis-free survival with a HR of 14.2 [31]. Nevertheless, no other validation studies have been conducted, and validated prognostic factors in stage II CC, such as the number of lymph nodes and MSI status, have not been included in the multivariate analysis. Five of these gene signatures are currently available: OncotypeDX®, GeneFx® Colon, ColoPrint®, OncoDefender-CRC®, and ColonPRS® (Table 1). Similarly, microRNA (miRNA) microarrays have been used to develop prognostic signatures in stage II CC [33, 36]. A six-miRNA classifier (which includes miR-21-5p, miR-20a-5p, miR-103a-3p, miR-106b-5p, miR-143-5p, and miR-215) clearly differentiated patients at high risk of relapse with a HR of 3.70 (95 % CI, 2.56–5.35) [33].
Relevant data have been published over the last years regarding the prognostic impact of these signatures; thus, it is important to evaluate its potential translation into the clinics.
Oncotype DX® (Genomic Health, Inc.)
OncotypeDx® is a quantitative multi-gene, real-time polymerase chain reaction assay developed in FFPE tumor samples from patients included in four clinical studies [34]. Initially, the signature comprised 18 genes including a set of genes to predict survival and another panel of genes to predict chemotherapy response. Since the subsequent validation study failed to demonstrated ability to predict treatment response, the signature was reduced to a recurrence risk score (RS), defined according to the expression of twelve genes. Three groups of patients were differentially identified according to the RS: low risk (RS < 30), intermediate risk (RS 30–40) and high risk (RS >/= 41). In the first validation set, which included patients from the QUASAR trial, the RS was able to separate patients randomized to surgery arm with high risk of recurrence (RR) and low RR (High RS, 3-year RR of 22 % and low RS, 3-year RR of 12 %) [7]. Moreover, its prognostic value was retained when other prognostic factors, such as T4 and microsatellite instability, were included in the multivariate analysis (HR = 1.43, p value = 0.006). These results have been further validated in two other clinical trials [37, 38•]. In the CALGB 9581 trial, (n = 690 stage II evaluable patients) the RS was associated with recurrence after tumor resection beyond mismatch repair, T stage, number of lymph nodes examined, histological grade, and lymphovascular invasion (HR = 1.68; 95 % CI, 1.18–2.38) [37]. Interestingly, the prognostic value of RS was most evident in the subgroup of T3 MSS patients: 5-year RR in the prespecified low and high RS groups were 13 % (95 % CI, 10 %–16 %) and 21 % (95 % CI, 16 %–26 %), respectively. In the NSABP C-07 trial, stages II and III CC patients were randomly assigned to FU or FU + oxaliplatin adjuvant chemotherapy. The RS demonstrated again an association with RR as well as with DFS and overall survival (OS) [38•]. Moreover, the additional benefit of oxaliplatin treatment was higher in patients with high RS, independently of TNM stage. It is noteworthy that the addition of oxaliplatin did not report an increase in DFS and OS in stage II in the full cohort of the study [39], but important differences were observed when the RS was applied. In stage II CC patients with low RS, there was not a positive effect of oxaliplatin addition (5-year RR of 7 % in the FU arm versus 12 % in the FU + oxaliplatin arm), whereas patients with high RS did benefit of oxaliplatin treatment with an absolute decrease in 5-year RR of 14 %. In stage III, similar results were observed. Although the RS cannot be considered a predictor of oxaliplatin treatment efficacy, it could discriminate in combination with stage patients with a higher RR; thus, a higher absolute benefit of oxaliplatin-based regimen can be derived.
In an attempt to evaluate the impact of RS results on physician recommendations regarding adjuvant chemotherapy in T3 pMMR stage II CC patients, a multicenter prospective study was conducted [40•]. A higher proportion of dMMR tumors (25 %) was observed compared with other large trials (14 % in QUASAR, 21 % in CALGB 9581), and 71 % of the patients were in the low risk group. In the primary analysis, treatment recommendations changed for 45 % (95 % CI, 36–53 %) of T3 pMMR patients after the RS results. Risk score result was also significantly associated with changes in treatment recommendation in the overall evaluable (pMMR and dMMR) population (p < 0.001).
Prognostic gene signatures have been mainly tested in CC cohorts. This prompted the performance of a validation study of the RS assay in stages II and III rectal cancer [41]. FFPE tumors from 297 patients randomized to surgery alone in the DUTCH TME trial were available for the analysis. Patients classified into the high RS group had higher RR than the low group in stage II (HR = 5.81, 95 % CI 2.33–14.50) but not in stage III, although it was not associated with shorter DFS and OS. In the multivariable analysis adjusted for clinicopathological factors, including radial margin status, the RS was a significant predictor of RR in stage II and stage IIIA/B. Consistency of the results from this study with those previously performed in CC cohorts supports the existence of common biological factors associated with prognosis, in accordance with data describing no major transcriptomic and genomic differences between colon and rectal tumors [32, 35].
GeneFx® Colon (Precision Therapeutics, Inc.)
GeneFx® Colon is a 634-transcript DNA microarray-based gene signature developed using FFPE tumor samples of 215 stage II CC patients [9]. In the first validation study, the signature was able to discriminate patients with higher relapse rate and cancer-related death with a HR of 2.53 (p value <0.001) and 2.21 (p value = 0.0084), respectively. The prognostic value remained when conventional pathological risk factors were added in the multivariate analysis (HR = 2.551; 95 CI, 1.471–4.423). Furthermore, results from data derived from patients included in the CALGB 9581 adjuvant trial has been recently communicated [42]. In this validation study, high-risk patients exhibited worse recurrence-free interval (HR = 2.0; 95 % CI, 1.3–3.3). The signature remained significant after adjustment for other prognostic factors, including MSI (HR = 2.1; 95 % CI, 1.3–3.4).
ColoPrint® (Agendia NV)
ColoPrint® is an 18-gene signature developed in a cohort of 188 stages I–IV CRC patients which classifies patients into high risk and low risk for distant metastasis recurrence [8]. An independent validation set including stages II and III CC patients demonstrated a significant association with relapse-free survival (RFS). Patients classified as low risk had a 5-year RFS rate of 87.6 % whereas high risk patients had a 5-year RFS rate of 67.2 % (p value = 0.003). In multivariate analysis, the signature remained one of the most significant prognostic factors, with a HR of 2.69 (95 % CI, 1.41–5.14). The prognostic value was most evident in the subgroup of stage II CC patients, even after the inclusion of the ASCO risk criteria in the analysis. In a pooled analysis of 416 stage II CC patients, ColoPrint® identified 63 % of patients as low risk [43•]. Risk of recurrence was significantly higher in the high risk group (5-year RR, 20.9 % versus 10.3 %, p value = 0.004). In the multivariate analysis, including number of lymph nodes resected and MSI status, the signature was the only factor that remained prognostic (HR = 2.16; 95 % CI, 1.28–3.65). An additional subgroup analysis was performed including patients with T3 and microsatellite stable tumors. Interestingly, the signature retained its prognostic value and patients classified as high risk had 2.4-fold higher RR than those classified as low risk. It should be pointed out that in this pooled dataset, risk classification using the NCCN guidelines was unable to separate high- and low-risk patients. Again, ColoPrint® demonstrated an improved accuracy in stage II risk classification beyond classical clinicopathological factors.
A prospective multicenter trial, the Prospective Analysis of Risk Stratification by ColoPrint (PARSC) trial, has included more than 500 stage II CC patients, and was designed to validate the performance of ColoPrint® in estimating the 3-year relapse rate in patients with stage II CC [44]. Besides, this study also pretends to analyze the impact of ColoPrint® on adjuvant treatment decision (ClinicalTrials.gov Identifier: NCT00903565).
OncoDefender-CRC® (Everist Genomics, Inc.)
OncoDefender-CRC® is a 5-gene expression assay developed from FFPE tissues of stage I and II CRC patients [45]. In the external validation analysis, the signature was capable of distinguishing patients at high risk from those at low risk for recurrence within 3 years after curative resection with a HR of 1.80 (95 % CI, 1.19–2.71). Besides, the signature was compared with the National Comprehensive Cancer Network (NCCN) Guidelines. This comparison demonstrated that OncoDefender-CRC® performs better in terms of RR that NCCN Guidelines with a HR of 1.76 (p value = 0.013) and 0.897 (p value = 0.648), respectively. It is remarkable that 83 % of the patients classified as high risk because of insufficient lymph node resection (NCCN criteria) never recurred, suggesting that the need to retrieve more than 12 lymph nodes for accurate prognostication could be obviated. Although this 5-gene prognostic signature is commercially available, it is necessary to conduct prospective clinical trials, including the analysis of MSI status, to validate its utility and possible use in clinical routine.
ColonPRS® (Signal Genetics LLC)
Gene expression data from 232 stage I-IV CC patients identified a 163-probe set significantly associated with outcome [46]. Afterwards, it was validated in an independent series of stages II and III CC and clearly distinguished two groups of patients depending on the risk of recurrence (5-year DFS, HR of 3.19, p value = 0.021). However, in the multivariate analysis, the classifier was an independent prognostic factor in the training set only, maybe because of the small sample size of the validation set. None of the stage II CC patients classified as low risk relapsed after 5-years of follow-up. Despite this gene signature is available for clinical testing, there is a need to validate its clinical utility in larger prospective well-characterized series.
In an attempt to compare the prognostic value of different gene classifiers when applied to the same population, Di Narzo et al. evaluated four different risk scores in a prospective cohort [47]. FFPE tumor samples from 688 patients enrolled into the PETACC 3 trial were available for testing for Oncotype Dx®, Veridex [31], GeneFx® Colon, and the MDA prognostic signature [48•]. Although the four gene signatures presented little overlap, Oncotype Dx®, Veridex, and GeneFx® Colon had similar predictive value in terms of relapse-free survival (HR close to 1.30) even when adjusting for risk factors such as T-stage, N-stage, or MSI status. It is noteworthy that the RS defined by the different signatures concurred poorly, probably because each one is enriched with genes associated with different pathways that identify distinct subgroups of patients with increased risk of recurrence. Finally, a combined RS (including the average of the four scores derived from the different signatures) identified significantly better a subgroup of patients with a higher risk of recurrence and death (HR of 1.56 and 1.74, respectively).
It is worth pointing out that different CRC subtypes have been recently defined by means of unsupervised clustering methodology [49]. The CRC Subtyping Consortium proposed a new taxonomy for CRC which integrates gene expression-based classification and other biological characteristics, such as mutations, copy number aberrations (CNAs), or DNA methylation. Four distinct molecular subtypes could be identified: CMS1 (MSI immune), characterized by hypermutation, MSI and an strong immune activation; CMS2 (canonical), characterized by high prevalence of somatic CNAs and WNT and MYC signaling activation; CMS3 (metabolic), characterized by metabolic dysregulation and KRAS mutations; and CMS4 (mesenchymal), with high stromal infiltration and transforming growth factor-β activation. Moreover, this molecular characterization is clearly associated with clinical variables and prognosis, independently of classical clinicopathological and molecular prognostic markers. This comprehensive classification should be adopted by the community for CRC stratification in future clinical trials and for the development of new therapeutic strategies (Fig. 1).

The molecular colorectal cancer classification according to the Colorectal Cancer Subtyping Consortium and its potential role in the clinics. The new taxonomy of colorectal cancer based on unsupervised gene signatures, which also integrates distinct biological characteristics (mutations, copy number aberrations, DNA methylation, microRNA, and protein expression), have defined potential driver events associated with prognosis and/or benefit to therapy. This classification is clinically relevant and should be adopted for future pre-clinical and clinical research to develop novel-targeted drugs and other therapeutic strategies. CMS consensus molecular subtype, RCT randomized clinical trial
Clinical Implications
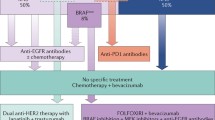
Fluoropyrimidine-based chemotherapy is routinely used after surgical resection in stage III CC as it has been demonstrated to improve survival. Nevertheless, there is insufficient data to support the administration of chemotherapy in stage II CC indiscriminately. Expert consensus suggests there is a potential benefit of adjuvant treatment in a subgroup of patients, characterized by clinicopathological features, with high risk of recurrence [5, 12]. Recently, microsatellite instability has been integrated into the algorithm of treatment decision in stage II CC, as long as patients with deficient MMR tumors have better survival. Moreover, the impact of microsatellite status on chemotherapy benefit in the adjuvant setting is controversial, although it is suggested that fluoropyrimidine-based chemotherapy does not improve survival [21, 50]. In this context, clinicians do not recommend adjuvant therapy in those patients with MSI stage II completely resected CC (Fig. 2). The prognostication of other molecular factors, mainly KRAS and BRAF, are still debatable due to lack of conclusive data. Nevertheless, results from randomized clinical trials in stage III CC pointed out that KRAS/BRAF mutations significantly decreased survival in the subgroup of patients with MSS tumors, although this could be more evident depending on the location. In conclusion, KRAS or BRAF may influence outcome in a subgroup of localized CC patients but more evidence is required to establish the mutational analysis in clinical routine for risk stratification. If KRAS/BRAF mutational status is related with stage II CC prognosis and if it should be considered for the administration of adjuvant treatment remain unknown.

Algorithm for adjuvant chemotherapy decision. MSI microsatellite instability, MSS microsatellite stable
The utility of gene expression signatures as prognostic tools in CRC patients has been largely evaluated. It is important to highlight that the applicability of these signatures depends on the efficacy in prediction and technological issues, mainly. In clinics, most of tissue samples collected during surgery as well as biopsies, are fixed and embedded in paraffin, thus FFPE samples are the most widely available source of tissue material for genomic studies. It is well known that microarray analysis with FFPE has technical limitations due to RNA degradation. The advances in methodological and technological aspects have allowed the development of high-quality RNA signatures from FFPE specimens. Several classifiers developed in training sets including fresh colorectal tumor samples, but only few of them have been validated in independent and external cohorts of stages II and III CC with FFPE tumor samples. Oncotype DX®, GeneFx® Colon, and ColoPrint® emerged as the candidate prognostic gene expression classifiers for the foreseeable future because of the robustness of their role in risk assessment. Several issues need to be considered before introducing new technology and assays into the clinics [51••]. It is mandatory to compare gene profiles with established risk factors, including MSI status. Accordingly, Oncotype DX®, GeneFx® Colon, and ColoPrint® demonstrated their capability to improve standard risk classification in large and prospective validation studies. Moreover, both Oncotype DX® and ColoPrint® discriminated better in the T3MSS subgroup of patients, which is clinically most relevant. Although gene expression signatures are not recommended in clinical practice, clinicians should consider its use in T3MSS CC patients for avoiding unnecessary adjuvant chemotherapy in those patients classified as low risk (Fig. 2).
Conclusions
Adjuvant therapy in stage III CC is still the standard of care, independently of the recent data regarding the negative impact on survival of KRAS and BRAF mutations. In stage II CC, there is still insufficient data supporting the use of adjuvant chemotherapy. Nevertheless, a subgroup of patients at higher risk of recurrence characterized by clinicopathological factors, MSI status, and gene profiling should be considered for adjuvant fluoropyrimidine-based therapy. Clinical trials if available must always be taken into account in this setting. Moreover, further investigation in different molecular pathways involved in CC as well as greater external validity and consensus among different gene expression platforms are required to improve clinical practice in early stage of CC.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ferlay J et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2011;127(12):2893–917.
Andre T et al. Phase III study comparing a semimonthly with a monthly regimen of fluorouracil and leucovorin as adjuvant treatment for stage II and III colon cancer patients: final results of GERCOR C96.1. J Clin Oncol. 2007;25(24):3732–8.
Andre T et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27(19):3109–16.
Benson 3rd AB et al. Localized colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Cancer Netw. 2013;11(5):519–28.
Labianca R et al. Primary colon cancer: ESMO Clinical Practice Guidelines for diagnosis, adjuvant treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v70–7.
Santos C et al. Molecular markers in colorectal cancer: clinical relevance in stage II colon cancer. Colorect Cancer. 2013;2(3):1–21.
Gray RG et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29(35):4611–9.
Salazar R et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2010;29(1):17–24.
Kennedy RD et al. Development and independent validation of a prognostic assay for stage II colon cancer using formalin-fixed paraffin-embedded tissue. J Clin Oncol. 2011;29(35):4620–6.
Sargent DJ et al. Molecular testing for lymph node metastases as a determinant of colon cancer recurrence: results from a retrospective multicenter study. Clin Cancer Res. 2014;20(16):4361–9.
Samowitz WS et al. PCR versus immunohistochemistry for microsatellite instability. J Mol Diagn. 2008;10(2):181–2. author reply 181.
NCCN Guidelines. Colon Cancer. Version 3 2015. https://www.nccn.org.
Bokemeyer C et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–75.
Douillard JY et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–705.
Van Cutsem E et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33(7):692–700.
Douillard JY et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34.
Taieb J et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(8):862–73.
Sinicrope FA et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31(29):3664–72.
Yoon HH et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in BRAF-wild-type stage III colon cancers from an adjuvant chemotherapy trial (N0147 alliance). Clin Cancer Res. 2014;20(11):3033–43.
Taieb J, et al. Prognostic value of BRAF V600E and KRAS exon 2 mutations in microsatellite stable (MSS), stage III colon cancers (CC) from patients (pts) treated with adjuvant FOLFOX+/- cetuximab: A pooled analysis of 3934 pts from the PETACC8 and N0147 trials. J Clin Oncol. 2015;33 suppl:abstr 3507.
Hutchins G et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29(10):1261–70.
Peeters M et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19(7):1902–12.
Oliner KS, Douillard JY, et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first-line treatment (tx) for metastatic colorectal cancer (mCRC). J Clin Oncol. 2013;31 suppl:abstr 3511.
Roth AD et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2009;28(3):466–74.
Pietrantonio F et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51(5):587–94.
Van Cutsem E et al. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;25 Suppl 3:iii1–9.
Sinicrope FA et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2014;148(1):88–99.
Sanz-Pamplona R et al. Clinical value of prognosis gene expression signatures in colorectal cancer: a systematic review. PLoS One. 2012;7(11):e48877. A systematic review of the gene expression signatures evaluated as pronostic tools in colon cancer.
Sveen A et al. Anticipating the clinical use of prognostic gene expression-based tests for colon cancer stage II and III: is Godot finally arriving? Clin Cancer Res. 2013;19(24):6669–77.
Wang Y et al. Gene expression profiles and molecular markers to predict recurrence of Dukes’ B colon cancer. J Clin Oncol. 2004;22(9):1564–71.
Jiang Y et al. Development of a clinically feasible molecular assay to predict recurrence of stage II colon cancer. J Mol Diagn. 2008;10(4):346–54.
Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330-7.
Zhang JX et al. Prognostic and predictive value of a microRNA signature in stage II colon cancer: a microRNA expression analysis. Lancet Oncol. 2013;14(13):1295–306.
Kerr D et al. A quantitative multigene RT-PCR assay for prediction of recurrence in stage II colon cancer: Selection of the genes in four large studies and results of the independent, prospectively designed QUASAR validation study. J Clin Oncol (Meeting Abstracts). 2009;27(15S):4000.
Sanz-Pamplona R et al. Gene expression differences between colon and rectum tumors. Clin Cancer Res. 2011;17(23):7303–12.
Schepeler T et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416–24.
Venook AP et al. Biologic determinants of tumor recurrence in stage II colon cancer: validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J Clin Oncol. 2013;31(14):1775–81.
Yothers G et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol. 2013;31(36):4512–9. The largest validation study of OncotypeDx in a patient population included in a randomized clinical trial.
Yothers G, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol. 2011;29(28):3768–74.
Srivastava G et al. Prospective multicenter study of the impact of oncotype DX colon cancer assay results on treatment recommendations in stage II colon cancer patients. Oncologist. 2014;19(5):492–7. The only clinical trial evaluating the impact of the gene expression signature results on treatment decision in stage II colon cancer.
Reimers MS, et al. Validation of the 12-gene colon cancer recurrence score as a predictor of recurrence risk in stage II and III rectal cancer patients. J Natl Cancer Inst. 2014;106(11):1–8.
Niedzwiecki D, et al. Association between ColDx assay result and recurrence-free interval in stage II colon cancer patients on CALGB (Alliance) 9581. J Clin Oncol. 2014;32 suppl 3:abstr 455.
Kopetz S et al. Genomic classifier ColoPrint predicts recurrence in stage II colorectal cancer patients more accurately than clinical factors. Oncologist. 2015;20(2):127–33. Results derived from a pooled dataset of stage II colon cancer which validates the prognostic value of ColoPrint.
Salazar R, et al. Comparison of ColoPrint risk classification with clinical risk in the prospective PARSC trial. J Clin Oncol. 2014;32(5s, suppl; abstr 3562).
Lenehan PF, et al. Generation and external validation of a tumor-derived 5-gene prognostic signature for recurrence of lymph node-negative, invasive colorectal carcinoma. Cancer. 2012;118(21):5234–44.
Van Laar RK. An online gene expression assay for determining adjuvant therapy eligibility in patients with stage 2 or 3 colon cancer. Br J Cancer. 2010;103(12):1852–7.
Di Narzo AF et al. Test of four colon cancer risk-scores in formalin fixed paraffin embedded microarray gene expression data. J Natl Cancer Inst. 2014;106(10). Study analysing the prognostic value of four different classifiers in the same cohort of patients.
Oh SC et al. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut. 2012;61(9):1291–8.
Guinney, J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015.
Des Guetz G et al. Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer. 2009;45(10):1890–6.
Salazar R, Tabernero J. New approaches but the same flaws in the search for prognostic signatures. Clin Cancer Res. 2014;20(8):2019–22. Critical overview of the steps for developing and validating prognostic multigene classifiers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Personalized Medicine in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Vivas, C.S., Sanz-Pamplona, R., Grasselli, J. et al. Are Gene Signatures Ready for Use in the Selection of Patients for Adjuvant Treatment?. Curr Colorectal Cancer Rep 12, 18–26 (2016). https://doi.org/10.1007/s11888-016-0305-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-016-0305-x