
Abstract
Purpose of Review
There has been much debate surrounding novel medical therapies and heart transplantation listing challenges in patients with hypertrophic cardiomyopathy (HCM).
Recent Findings
Recent clinical trials led to FDA approval of mavacamten (a cardiac myosin inhibitor), offering symptom relief and potentially delaying/avoiding invasive septal reduction therapies for some patients with HCM and left ventricular outflow obstruction (LVOTO). For those with refractory symptoms and end-stage heart failure, heart transplantation remains the gold standard. However, the concern for the organ allocation system failing to prioritize those individuals persists.
Summary
HCM is a heterogeneous genetic condition with variable penetration and clinical presentation. Even though a large portion of patients remain asymptomatic, an important minority develops debilitating symptoms refractory to medical therapy. Post-HT short- and long-term outcomes are favorable. However, HT waitlist mortality remains high. For highly selected patients with HCM, a left ventricular assist device is a viable option.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM) is defined by the presence of left ventricular hypertrophy (LVH) in the absence of other potentially causative cardiac, metabolic, or systemic conditions [1]. It is the most common inheritable monogenic cardiac disease with prevalence ranging from 1:1,124 to 1:200 [2,3,4,5]. HCM is considered an entity caused by mutations in one of at least 11 genes encoding sarcomeric or Z disc proteins. The most common pathogenic gene variants are myosin-binding protein C3 (MYBPC3) and myosin heavy chain 7 (MYH7) [6,7,8]. Given that HCM can be inherited as a Mendelian autosomal dominant disorder, it is estimated that in the United States alone there are at least 750,000 individuals affected by this condition. The fact that only about 100,000 patients are carrying this diagnosis, suggests that HCM is significantly underdiagnosed [9].
The condition now recognized as HCM was first reported by French clinicians between 1868–1869 and subsequently by French pathologist Dr. Teare in 1958 [10,11,12,13]. Shortly after the latter, its clinical description was presented by the Braunwald group from the National Institutes of Health [14]. This inaugurated an era of clinical research that revealed this condition to have variable penetrance and a wide range of clinical presentations.
Clinical manifestation and diagnosis
Many individuals with HCM experience no or only mild exercise intolerance. However, an important minority suffers from debilitating symptoms such as chest pain, syncope, and dyspnea on exertion. Those can be attributed to a range of pathophysiologic mechanisms such as LV outflow tract obstruction, mitral regurgitation, heart failure with preserved or reduced ejection fraction (HFpEF or HFrEF, respectively), and atrial or ventricular arrhythmias [1, 15,16,17,18,19,20,21,22,23,24]. In some cases sudden cardiac death (SCD) is the first manifestation of the disease.
Transthoracic echocardiography (TTE) is the initial and preferred imaging modality used for making an HCM diagnosis. Cardiac magnetic resonance imaging (cMRI) is also indicated and particularly useful when TTE fails to provide optimal images or is inconclusive (i.e. borderline LVH or concern for apical HCM). For patients who cannot have a cMRI performed, cardiac computer tomography (CCT) can be considered. Regardless of the imaging modality used, end-diastolic wall thickness ≥ 15 mm in the absence of other potential causes of hypertrophy in a non-dilated LV is consistent with the HCM diagnosis. If there is a family history of HCM or a known genetic mutation, a wall thickness of ≥ 13 mm can be diagnostic [1].
There are several hypertrophy patterns seen in patients with HCM. The most commonly observed is asymmetric septal hypertrophy (also called a reversed septal curvature) [25]. In up to 70% of patients, hypertrophied septum protrudes into the LV cavity and results in obstructive physiology. Within this cohort, resting left ventricular outflow obstruction (LVOTO) defined by an LV intracavitary peak gradient ≥ 30 mmHg is seen at rest in 25–30% of patients [26, 27]. In another 35% LVOTO is inducible only by provocative maneuvers (i.e. Valsalva or amyl nitrate inhalation). If those fail to evoke LVOTO, the exercise echocardiography is recommended [27, 28]. The presence of a resting or provoked gradient ≥ 30 mmHg is associated with an increased risk of progression to New York Heart Association (NYHA) class III or IV HF symptoms and SCD [29]. The remaining 30% of patients with HCM have LV outflow gradient < 30 mmHg at rest and/or with exercise, and are classified as having non-obstructive HCM [26, 30, 31]. One of the anatomical patterns of hypertrophy with non-obstructive physiology is apical HCM. It is characterized by mid and apical segment hypertrophy and a spade-shaped LV cavity seen on imaging [25].
Ventricular arrythmias
Patients with HCM are at risk of SCD in the mechanism of ventricular tachycardia (VT) and ventricular fibrillation (VF). Antiarrhythmics and catheter-based ablation do not reduce the risk of ventricular tachyarrhythmias in this cohort, making an implantable cardioverter-defibrillator (ICD) the only currently available effective treatment. Although ICDs revolutionized the landscape of ischemic heart disease in the 1980s, it was not until a sentinel New England Journal of Medicine publication by Maron et al. in 2000, that they were incorporated into the management of patients with HCM [32,33,34]. That contributed to a substantial reduction in the yearly mortality within this group, from 6–8% to 0.5% regardless of gender, race, or age. Notably, none of the medications used in patients with HCM has proven to reduce their mortality [35].
Obstructive HCM treatment
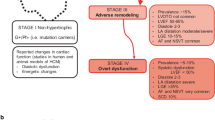
Pharmacotherapy in HCM is reserved for symptomatic patients only (Fig. 1). In 1964, β-blockers were introduced by Dr. Braunwald to alleviate HCM symptoms and they constitute a first-line HCM therapy in the current era as well [14]. The advent of β-blockers was followed by non-dihydropyridine calcium channel blockers (CCBs), predominantly verapamil, which are used as the second-line therapy [36,37,38]. If β-blockers and CCBs fail to control LVOTO symptoms disopyrimide can be added. It is a class Ia antiarrhythmic with potent negative inotropic properties and was the first agent proven to reduce LVOTO at rest (by 50%) serving as an option to delay elective myectomy (or alcohol septal ablation) [37]. However, its use is often limited by significant anticholinergic side effects.

Therapeutics for Hypertrophic Cardiomyopathy Across the Disease Spectrum
In recent years cardiac myosin inhibitors (CMIs) have been becoming the preferred medical therapy in HCM with LVOTO, with mavacamten being the first-in-class Food and Drug Administration-approved agent [39, 40]. EXPLORER-HCM (Evaluation of Mavacamten in Adults With Symptomatic Obstructive Hypertrophic Cardiomyopathy) and MAVA-LTE (A Long-Term Safety Extension Study of Mavacamten in Adults Who Have Completed EXPLORER-HCM) clinical trials demonstrated that mavacamten more frequently than placebo resulted in reduction in a resting LVOT gradient to < 30 mm Hg along with at least one NYHA functional class improvement and an increase of peak VO2 ≥ 1.5 mL/kg/min or an improvement of peak VO2 by 3.0 mL/kg/min without compromising the baseline NYHA class [41]. The agent was well tolerated overall. In 6% of trial participants a reversible reduction in left ventricular ejection fraction (LVEF) to < 50% was observed [42]. Therefore, the mavacamten dose should be titrated under echocardiographic surveillance of LVEF [43]. In 2023, the European Society of Cardiology updated their cardiomyopathy management guidelines and recommended considering (1) adding mavacamten to a β-blocker to alleviate the symptoms in patients with HCM and LVOTO at rest or with exercise (class IIa, level A) (2) using it in monotherapy in patients with LVOTO at rest or with exercise who are intolerant of or have contraindications to β-blocker, CCB, or disopyrimide-based therapy (class IIa, level B) [43]. In 2024, the AHA/ACC guidelines for the management of HCM included mavacamten in the medical treatment algorithm, recommending its use in patients with symptoms refractory to at least one of the first-line agents (β-blockers or CCB) [1]. The subsequent clinical trial, VALOR-HCM (A Study to Evaluate Mavacamten in Adults With Symptomatic Obstructive HCM Who Are Eligible for Septal Reduction Therapy), demonstrated that the administration of mavacamten as part of maximally tolerated medical therapy resulted in a lower need for invasive septal reduction therapies compared to a placebo group [44]. Since mavacamten mitigates the progression of HF symptoms, in the future, it might be indicated in patients who do not have access to an experienced center performing septal myectomy or alcohol ablation or those wishing to delay/avoid it [45,46,47].
The next promising agent in the CMI group is aficamten. Results from the SEQUOIA-HCM (Safety, Efficacy, and Quantitative Understanding of Obstruction, Impact of Aficamten in HCM) clinical trial, announced at the Heart Failure Association (HFA) conference in Lisbon in May 2024, showed that after 24 weeks of aficamten treatment in patients with LVOTO, there was a significantly greater improvement in peak myocardial oxygen consumption (pVO2) compared to the placebo group. The average change in pVO2 in the treated group was 1.8 ml/kg/min (95% confidence interval [CI]: 1.2 to 2.3). Additionally, an improvement of at least one NYHA functional class occurred more freuqently in the treatment than in the placebo group (58.5% vs. 24.3%, p < 0.01). Reduction of LVOT gradient to < 30 mm Hg after Valsalva maneuver was seen in 49.3% and 3.6% of aficamten and placebo patients respectively. The frequency of adverse events was similar in both groups. Aficamten has an advantage over mavacamten: it reaches pharmacokinetic steady state more quickly (2 weeks compared to 6 weeks for mavacamten) and does not induce cytochrome P450 (CYP) enzymes, thereby reducing the risk of drug-to-drug interactions [48].
Septal reduction therapy
For most patients with obstructive HCM, LVOT gradient > 50 mm Hg at rest or with exercise and NYHA class III/IV symptoms refractory to optimal medical therapy, transaortic septal myectomy is the preferred treatment method [1]. It results in immediate relief of outflow obstruction, reduction in left ventricular end-diastolic pressure (LVEDP), and improvement in symptoms, while preserving systolic function. When performed at highly-volume HCM centers with highly experienced surgeons, myectomy is associated with the most favorable outcomes and lowest perioperative complications (high-degree heart block requiring a permanent pacemaker 1–5%, mortality 0.5%) [49]. Some surgeons successfully combine septal myectomy with simultaneous mitral valve apparatus repair and intraoperative ablation, which reduces the risk of heart failure symptoms and atrial fibrillation recurrence [50, 51].
For patients ineligible for septal myectomy, percutaneous alcohol septal ablation (ASA) has become the most frequent alternative. Similar to septal myectomy, this procedure is also recommended to be performed at a high-volume HCM center by an interventionalist experienced with the morphological variability of this entity. Even though both recovery and hospital stay are shorter after ASA when compared to myectomy, the reduction in LV gradient can be variable and may not be immediate (> 90 days), and the incidence rate of complete heart block requiring a permanent pacemaker is higher (10%-15%). Moreover, in patients with extreme or only mild septal hypertrophy or those with mitral valve apparatus abnormalities, the results of ASA are inconsistent. There is also a small group of patients who develop ventricular arrhythmias as a result of alohol-induced septal scar [52].
Non-obstructive HCM treatment
Patients with HCM without LVOTO constitute a therapeutic challenge. Commonly observed in this cohort symptoms, dyspnea, and chest pressure/angina, result from increased LV filling pressures due to diastolic dysfunction, increased myocardial oxygen demand, and microvascular dysfunction [1]. Therefore, current therapies for HCM without LVOTO consist of β -blockers, verapamil, and diuretics. For patients with debilitating angina or HF symptoms refractory to medical therapy, heart transplantation is the only definite therapy (Fig. 1).
So far, there are no guidelines for the use of CMI in this group of patients. Results from the MAVERICK-HCM clinical trial (Mavacamten in Adults with Symptomatic Non-Obstructive Hypertrophic Cardiomyopathy) suggest that mavacamten may reduce myocardial wall tension in patients with HCM without LVOTO [53]. Currently, a phase 3 clinical trial (ODYSSEY-HCM: A Study of Mavacamten in Non-Obstructive Hypertrophic Cardiomyopathy; NCT05582395) is underway to determine whether mavacamten leads to symptom reduction, NYHA class improvement, and increased pVO2 in patients with HCM without LVOTO. The results are expected to be available in 2025.
Heart failure
Substantial clinical heterogeneity of HCM makes ascertaining the incidence of HF in this group of patients challenging. Therefore, it has been reported to be anywhere from 50 to 67% [26]. Those numbers can be overestimated given that a significant number of asymptomatic patients with HCM remain undiagnosed. HF has two distinct clinical presentations in patients with HCM. In the majority of them, it manifests as HFpEF affecting mostly patients with obstructive and about 10% with non-obstructive HCM [26]. Only a minority of patients develop HFrEF, also described as end-stage HCM. While the progress of HCM is not uniform, and HF symptoms can arise or worsen at any age, midlife is the most common timeframe of their onset [22, 33, 35, 37, 54]. HF symptoms constitute a predominant manifestation of HCM in women, who are usually referred to HCM centers at an older age, with higher LVOTO gradients and worse cardiopulmonary exercise testing (CPET) measures [54, 55].
Symptoms of HF frequently fluctuate (“good and bad days”) and are greatly dependent on LV preload [1, 35]. Acute HF is observed seldom and can be precipitated by arrhythmia (atrial fibrillation) or worsening of mitral regurgitation [26]. Discontinuation of negative inotropes (CCB, disopyramide and CMIs) is of paramount importance. Patients with LVEF < 50% and non-obstructive HCM should be treated with guideline-directed medical therapy (GDMT) for HFrEF (β-blockers, ACE-I, ARB, ARNI, MRA, SGLT2i) and diuretics to relieve congestion [56]. There is a scarcity of data supporting cardiac resynchronization therapy (CRT) in patients with HCM [57]. However, those with LBBB and LVEF < 50% should be considered for CRT [35, 58].
Progression to the end-stage HF is observed in 3.5–17% of patients with HCM, predominantly as a result of LVH and LVOTO [26]. The risk of developing advanced HF is 3.2% per year among those with obstructive and 1.6% per year among those with non-obstructive HCM [35]. Peak myocardial oxygen consumption assessed by CPET is one of the objective measures reflecting a patient's functional capacity and determining candidacy for advanced HF therapies. Its reduced value (< 14 ml/kg/min or < 50% predicted for age) correlates with the NYHA functional class in most, but not all, symptomatic patients with HCM. As demonstrated in a single-center study, up to 16% of patients with HCM requiring advanced HF therapies have pVO2 above that cutoff [20]. That showcases a significant mismatch between pVO2 and the actual level of functional limitations in patients with HCM. Therefore, this variable alone should not be used as an argument for excluding them from being considered for advanced HF therapies [59].
LVAD
The advent of left ventricular assist devices (LVADs) has substantially changed the treatment landscape for patients with advanced HF refractory to GDMT. The use of LVADs allowed not only to improve the quality of life but more importantly increased survival of patients with ischemic and dilated non-ischemic cardiomyopathy (ICM and NICM, respectively) [60, 61]. For patients with HCM the implementation of LVAD therapy has been more challenging, primarily due to anatomical differences compared to those with dilated cardiomyopathy. Patients with HCM usually have non-dilated LV cavities with thick fibrotic walls which can make LVAD implantation surgically challenging and post-operatively puts patients at risk of ongoing impaired diastolic filling, inflow cannula obstruction, and suction events [62, 63]. The combination of the latter two can propagate ventricular arrhythmias, lead to low pump flows and cannula thrombosis [62]. In patients with HCM and dilated end-stage phenotype, extensive ventricular remodeling with replacement fibrosis is observed. That results in increased ventricular stiffness and might contribute to continued diastolic dysfunction after LVAD implantation [63].
Individuals with HCM and end-stage HF have been usually excluded from LVAD clinical trials. As a result, the post-LVAD outcomes in this population are not well defined. Both the 2016 International Society for Heart and Lung Transplantation (ISHLT) guidelines and the 2024 American College of Cardiology/American Heart Association (ACC/AHA) guidelines on the diagnosis and management of HCM recommend heart transplantation and only selective use of LVADs for patients with HCM who develop HF refractory to medical therapy [1, 64]. However, a handful of single-center observational studies demonstrated that carefully selected patients with HCM have comparable outcomes to the NICM LVAD-supported group at 1- and 3-years (Table 1) [65, 66, 67]. Notably, all patients with HCM included in those studies had dilated end-stage phenotype with severely reduced LV systolic function and only moderate septal hypertrophy [62, 66, 68, 69]. Higher preoperative left ventricular end-diastolic diameter (LVEDD > 46 mm) was an important determinant of better post-LVAD mortality and morbidity [66, 70]. However, one study showcased that in patients with pre-operative LVEDD of 70 mm axial configuration LVAD was associated with a higher risk of device thrombosis compared to centrifugal configuration (HR 1.61, 95%CI 1.17–2.22, p < 0.01). This risk continued to rise as the LVEDD increased suggesting a U-shaped phenomenon with extreme LVEDD values being associated with inferior post-LVAD outcomes [69]. Additionally, right ventricular failure is of concern in this cohort. Its incidence post-LVAD implantation is observed in 12.5% to 50% of patients and is significantly higher than in ICM/NICM cohorts [62, 66, 67]. This can be attributed to a long-standing pulmonary hypertension and RV myocardial dysfunction beyond the obvious LV findings.
The largest to-date analysis of patients with HCM (n = 94) who underwent continuous-flow LVAD implantation (2008 and 2014) was performed using the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) (Table 1). HCM patients were younger which was consistent with previous single-center studies, and less frequently INTERMACS 1–3 compared to the NICM group. Although left ventricles of patients with HCM were dilated by standard criteria, they were smaller compared to those with NICM. Patients with LVEF < 40% constituted 95% of studied patients with HCM. These patients showcased LV cavity dilation and wall thinning consistent with dilated end-stage HCM phenotype that resembles NICM. The rate of patients with HCM with moderately or severely reduced RV function based on TTE findings was comparable to the NICM group. Survival rate at 1 year was similar between patients with HCM and NICM: 81.4% (50.2% alive on LVAD, 31.5% were transplanted) vs 81.7%. The rate of adverse events (arrhythmias, pump malfunction, hemolysis, CVA, renal dysfunction) was similar between the HCM and NICM groups. There was a clear effect of higher mortality in patients with smaller LV cavity (for pre-implantation LVEDD greater or smaller than 50 mm: 35% vs 88%, p < 0.01) [62].
In conclusion, in the current clinical practice, LVAD implantation can be considered for highly selected patients with HCM with LV cavity dilation and LV systolic dysfunction consistent with the dilated end-stage phenotype, and who have exhausted all other alternative therapeutic options.
Heart transplantation
Heart transplantation (HT) is the gold-standard treatment for end-stage HF in patients with HCM. In the early 1990s, few patients with non-obstructive HCM and HF refractory to medical therapy were referred for HT [64]. Since then the number of HTs performed in patients with HCM has been growing exponentially. Given that HCM is still a relatively rare disease, patients with HCM constitute about 2–3% of all HTs performed, and those with HFpEF comprise about one-half of all patients with HCM listed for HT [35, 71].
Reports on post-HT outcomes of patients with HCM are summarized in Table 2. Several single-center studies along with a national registry-based report demonstrated that patients with HCM undergoing HT compared to their non-HCM counterparts were younger (41–48 vs. 55–57 years) and had fewer comorbidities (diabetes, peripheral vascular disease, hypertension, hyperlipidemia, chronic kidney disease, and chronic obstructive pulmonary disease). A national registry-based study demonstrated that functional status at listing was better among patients with HCM compared to the ICM and NICM cohorts (62.1% vs 53.4% vs 52.3%, respectively, p < 0.01) [19, 72,73,74,75,76,77].
In 2015, a two-center small sample (n = 14) study reported higher 1-year post-HT mortality due to RV failure attributed to the presence of pulmonary hypertension [19]. Subsequent single-center publications demonstrated favorable 1-, 5-, and 10-year post-HT survival among recipients with HCM despite more challenging immediate post-HT course [19, 74, 77, 78]. Their survival was superior to that observed in recipients with ischemic cardiomyopathy and comparable to recipients with dilated non-ischemic cardiomyopathy [77]. The analysis of the United Network for Organ Sharing (UNOS) registry data demonstrated that the 1-, 5-, and 10-year survival among recipients with HCM was 85%, 75%, and 61%, respectively, and confirmed a trend of a better survival when compared to non-HCM recipients. (82%, 80%, and 49%, p = 0.05) [77]. Notably, 5-year survival reported by most individual HCM centers was > 90% [75]. More contemporary Scientific Registry of Transplant Recipients (SRTR)-based analysis demonstrated that survival in patients with HCM compared to the ICM cohort was significantly higher at 1 year (91.6% versus 87.5%; p = 0.03) with even more pronounced difference between those two groups at 5 years (82.5% versus 75.3%; p = 0.01). Survival of the NICM group was comparable [72]. None of the aforementioned studies demonstrated that the diagnosis of HCM was an independent predictor of post-HT mortality.
Even though post-HT outcomes in recipients with HCM are favorable, their waitlist mortality remains high [19]. Patients with HCM often do not benefit from inotropes as they can trigger ventricular arrhythmias. Those with small LV cavity and impaired filling are usually not eligible for durable mechanical circulatory support (MCS) as a bridge to transplantation which often results in organ allocation systems precluding them from being prioritized for HT [26]. To address these concerns, in 2018 a new 6-tiered organ allocation system was introduced in the US. Aside from prioritizing patients with temporary rather than durable MCS, it created an additional “exception” pathway for those whose pathophysiology, like in patients with HCM, does not fall into a typical realm of HFrEF. That resulted in higher transplant rates among patients with HCM without compromising their 1- and 3-year post-HT mortality. Nonetheless, waitlist mortality remained unchanged [79,80,81].
Conclusions
HCM is a heterogeneous condition with variable penetrance and non-uniform course. Contemporary therapies, particularly ICD, have significantly reduced sudden cardiac death-related mortality in this cohort. Recent clinical trials led to FDA approval of mavacamten (a cardiac myosin inhibitor), offering symptom relief and potentially delaying/avoiding invasive septal reduction therapies for some patients with HCM and left ventricular outflow obstruction LVOTO. Up to 17% of patients with HCM develop end-stage HF. Assessment of those patients needs to be based on a collection of variables (hemodynamics, CPET, and imaging) rather than a single data point. Although LVADs are not the optimal therapy for most individuals with HCM, they can be considered for highly selected patients with dilated end-stage phenotype if all possible alternative options have been exhausted. Heart transplantation remains the gold standard for patients with HCM and end-stage HF. The number of HTs performed in this cohort has been rising as the post-HT outcomes continue to be favorable. Nonetheless, designing an organ allocation system that would successfully reduce their waitlist mortality remains a challenge.
Data Availability
No datasets were generated or analysed during the current study.
References
Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. Writing Committee Members. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Am Coll Cardiol. 2024;83(23):2324–405.
Karim S, Chahal CAA, Sherif AA, Khanji MY, Scott CG, Chamberlain AM, et al. Re-evaluating the Incidence and Prevalence of Clinical Hypertrophic Cardiomyopathy: An Epidemiological Study of Olmsted County. Minnesota Mayo Clinic Proceedings. 2024;99(3):362–74.
Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–54.
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9.
Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol. 1999;33:1590–5.
Cirino AL, Harris S, Lakdawala NK, et al. Role of genetic testing in inherited cardiovascular disease: a review. JAMA Cardiol. 2017;2:1153–60.
Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–8.
Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–86.
Maron MS, Hellawell JL, Lucove JC, Farzaneh Far R, Olivotto I. Occurrence of clinically diagnosed hypertrophic cardiomyopathy in the United States. Am J Cardiol. 2016;117:1651–4.
Vulpian A. Contribution à l’étude des rétrécissements de l’orifice ventriculo-aortique. Arch Pathol. 1868;3:456–7.
Liouville H. Rétrécissement cardiaque sous aortique. Gazette Med Paris. 1869;24:161–3.
Hallopeau M. Rétrécissement ventriculo-aortique Gazette Med Paris. 1869;24:683–4.
Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20(1):1–8.
Braunwald E, Lambrew E, Rockoff D, et al. Idiopathic hypertrophic subaortic stenosis I. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30(Suppl IV):1–217.
Hutt E, Nissen SE, Desai MY. Unmet needs in the treatment of hypertrophic cardiomyopathy. Future Cardiol. 2021;17:1261–7.
Maron BJ, Rowin EJ, Casey SA, Maron MS. How hypertrophic cardiomyopathy became a contemporary treatable genetic disease with low mortality: shaped by 50 years of clinical research and practice. JAMA Cardiol. 2016;1:98–105.
Villemain O, Correia M, Mousseaux E, Baranger J, Zarka S, Podetti I, et al. Myocardial stiffness evaluation using noninvasive shear wave imaging in healthy and hypertrophic cardiomyopathic adults. JACC Cardiovasc Imaging. 2019;12:1135–45.
Soullier C, Obert P, Doucende G, Nottin S, Cade S, Perez-Martin A, et al. Exercise response in hypertrophic cardiomyopathy: blunted left ventricular deformational and twisting reserve with altered systolic-diastolic coupling. Circ Cardiovasc Imaging. 2012;5:324–32.
Pasqualucci D, Fornaro A, Castelli G, Rossi A, Arretini A, Chiriatti C, et al. Clinical spectrum, therapeutic options, and outcome of advanced heart failure in hypertrophic cardiomyopathy. Circ Heart Fail. 2015;8:1014–21.
Rowin EJ, Maron BJ, Carrick RT, Patel PP, Koethe B, Wells S, et al. Outcomes in patients with hypertrophic cardiomyopathy and left ventricular systolic dysfunction. J Am Coll Cardiol. 2020;75:3033–43.
Sorajja P, Nishimura RA, Gersh BJ, Dearani JA, Hodge DO, Wiste HJ, et al. Outcome of mildly symptomatic or asymptomatic obstructive hypertrophic cardiomyopathy: a long-term follow-up study. J Am Coll Cardiol. 2009;54:234–41.
Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114:2232–9.
Wilke I, Witzel K, Munch J, Pecha S, Blankenberg S, Reichenspurner H, et al. High incidence of de novo and subclinical atrial fibrillation in patients with hypertrophic cardiomyopathy and cardiac rhythm management device. J Cardiovasc Electrophysiol. 2016;27:779–84.
Hinojar R, Varma N, Child N, Goodman B, Jabbour A, Yu CY, et al. T1 mapping in discrimination of hypertrophic phenotypes: hypertensive heart disease and hypertrophic cardiomyopathy: findings from the international T1 multicenter cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2015;8:e003285.
Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by twodimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26:1699–708.
Seferovic PM, Polovina M, Bauersachs J, Arad M, Ben Gal T, Lund LH, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2019;21:553–76.
Nagueh SF, Phelan D, Abraham T, Armour A, Desai MY, Dragulescu A, et al. Recommendations for Multimodality Cardiovascular Imaging of Patients with Hypertrophic Cardiomyopathy: An Update from the American Society of Echocardiography, in Collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022;35:533–6.
Rowin EJ, Maron BJ, Olivotto I, Maron MS. Role of exercise testing in hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2017;10:1374–86.
Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295–303.
Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014;64:83–99.
Wigle ED. Cardiomyopathy: The diagnosis of hypertrophic cardiomyopathy. Heart. 2001;86:709–14.
Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342:365–73.
Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379:655–68.
Maron BJ, Nishimura RA, Maron MS. Shared decision-making in HCM. Nat Rev Cardiol. 2017;14:125–6.
Maron BJ, Rowin EJ, Udelson JE, Maron MS. Clinical spectrum and management of heart failure in hypertrophic cardiomyopathy. J Am Coll Cardiol HF. 2018;6:353–63.
Rosing DR, Kent KM, Maron BJ, Epstein SE. Verapamil therapy: a new approach to the pharmacologic treatment of hypertrophic cardiomyopathy: II, effects on exercise capacity and symptomatic status. Circulation. 1979;60(6):1208–13.
Sherrid MV, Barac I, McKenna WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45(8):1251–8.
Toshima H, Koga Y, Nagata H, Toyomasu K, Itaya K, Matoba T. Comparable effects of oral diltiazem and verapamil in the treatment of hypertrophic cardiomyopathy. Double-blind crossover study Jpn Heart J. 1986;27:701–15.
Hutt E, Desai MY. Medical Treatment Strategies for Hypertrophic Cardiomyopathy. Am J Cardiol. 2024;212:S33–41.
Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. Heart disease: a small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21.
Rader F, Choudhury L, Saberi S, Fermin D, Wheeler MT, Abraham TP, et al. Long-term safety of mavacamten in patients with obstructive hypertrophic cardiomyopathy: interim results of the Mavalong term extension (LTE) study. J Am Coll Cardiol. 2021;77:532–532.
Zampieri M, Berteotti M, Ferrantini C, Tassetti L, Gabriele M, Tomberli B, et al. Pathophysiology and Treatment of Hypertrophic Cardiomyopathy: New Perspectives. Curr Heart Fail Rep. 2021;18:169–79.
Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al. ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44:3503–626.
Desai MY, Owens A, Geske JB, Wolski K, Naidu SS, Smedira NG, et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol. 2022;80(2):95–108.
Maron BJ, Dearani JA, Smedira NG, Schaff HV, Wang S, Rastegar H, et al. Ventricular septal myectomy for obstructive hypertrophic cardiomyopathy (analysis spanning 60 years of practice): AJC Expert Panel. Am J Cardiol. 2022;180:124–39.
Maron MS, Ommen SR. Exploring new and old therapies for obstructive hypertrophic cardiomyopathy: mavacamten in perspective. Circulation. 2021;143:1181–3.
Maron BJ, Maron MS, Sherrid MV, Rowin EJ. Future role of new negative inotropic agents in the era of established surgical myectomy for symptomatic obstructive hypertrophic cardiomyopathy. J Am Heart Assoc. 2022;11: e024566.
Maron MS, Masri A, Nassif ME, Barriales-Villa R, Arad M, Cardim N, et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N Engl J Med. 2024 May 13. https://doi.org/10.1056/NEJMoa2401424. Online ahead of print.
Maron MS, Rastegar H, Dolan N, Carpino P, Koethe B, Maron BJ, Rowin EJ. Outcomes Over Follow-up ≥10 Years After Surgical Myectomy for Symptomatic Obstructive Hypertrophic Cardiomyopathy. Am J Cardiol. 2022;163:91–7.
Boll G, Rowin EJ, Maron BJ, Wang W, Rastegar H, Maron MS. Efficacy of combined CoxMaze IV and ventricular septal myectomy for treatment of atrial fibrillation in patients with obstructive hypertrophic cardiomyopathy. Am J Cardiol. 2020;125:120–6.
Stassano P, Di Tommaso L, Triggiani D, Contaldo A, Gagliardi C, Spampinato N. Mitral valve replacement and limited myectomy for hypertrophic obstructive cardiomyopathy: A 25-year follow-up. Tex Heart Inst J. 2004;31(2):137–42.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Management of Hypertrophic Cardiomyopathy JACC State-of-the-Art Review. J Am Coll Cardiol. 2022;79(4):390–414.
Ho CY, Mealiffe ME, Bach RG, Bhattacharya M, Choudhury L, Edelberg JM, et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–3266.
Rowin EJ, Maron MS, Wells S, Patel PP, Koethe BC, Maron BJ. Impact of sex on clinical course and survival in the contemporary treatment era for hypertrophic cardiomyopathy. J Am Heart Assoc. 2019;8:e012041.
Geske JB, Ong KC, Siontis KC, Hebl VB, Ackerman MJ, Hodge DO, et al. Women with hypertrophic cardiomyopathy have worse survival. Eur Heart J. 2017;38(46):3434–40.
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur. Heart J. 2014;35:2733–79.
Rogers DP, Marazia S, Chow AW, Lambiase PD, Lowe MD, Frenneaux M, et al. Effect of biventricular pacing on symptoms and cardiac remodelling in patients with end-stage hypertrophic cardiomyopathy. Eur J Heart Fail. 2008;10:507–13.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975.
Rowin EJ, Maron BJ, Kiernan MS, et al. Advanced heart failure with preserved systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant. Circ Heart Fail. 2014;7:967–75.
Miller LW, Pagani FD, Russell SD, et al. HeartMate II Clinical Investigators: Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med. 2007;357:885–96.
Slaughter MS, Rogers JG, Milano CA, et al. HeartMate II Investigators: Advanced heart failure treated with continuousflow left ventricular assist device. N Engl J Med. 2009;361:2241–51.
Patel SR, Saeed O, Naftel D, Myers S, Kirklin J, Jorde UP, Goldstein DJ. Outcomes of Restrictive and Hypertrophic Cardiomyopathies After LVAD: An INTERMACS Analysis. J Cardiac Fail. 2017;23(11):859–67.
Levine A, Gupta CA, Gass A. Advanced heart failure management and transplantation. Cardiol Clin. 2019;37:105–11.
Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, et al. International Society for Heart Lung Transplantation (ISHLT) Infectious Diseases, Pediatric and Heart Failure and Transplantation Councils. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35(1):1–23.
Yagi N, Seguchi O, Mochizuki H, Kuroda K, Nakajima S, Watanabe T, et al. Implantation of ventricular assist devices in hypertrophic cardiomyopathy with left ventricular systolic dysfunction. ESC Heart Failure. 2021;8:5513–22.
Topilsky Y, Pereira NL, Shah DK, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail. 2011;4:266–75.
Grupper A, Park SJ, Pereira NL, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: Improving outcomes for a lethal disease. J Heart Lung Transplant. 2015;34:1042–9.
Sridharan L, Wayda B, Truby LK, Latif F, Restaino S, Takeda K, et al. Mechanical Circulatory Support Device Utilization and Heart Transplant Waitlist Outcomes in Patients With Restrictive and Hypertrophic Cardiomyopathy. Circ Heart Fail. 2018;11(3):e004665.
Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the endstage phase of hypertrophic cardiomyopathy. Circulation. 2006;114:216–25.
Shah P, Birk S, Maltais S, Stulak J, Elmi A, Pagani FD, Cowger JA. Left ventricular assist device outcomes based on flow configuration and pre-operative left ventricular dimension: An Interagency Registry for Mechanically Assisted Circulatory Support Analysis. J Heart Lung Transplant. 2017;36:640–9.
Lund LH, Edwards LB, Dipchand AI, Goldfarb S, Kucheryavaya AY, Levvey BJ, et al. The registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Heart Transplantation Report-2016; focus theme: primary diagnostic indications for transplant. J Heart Lung Transplant. 2016;35:1158–69.
Zuñiga Cisneros J, Stehlik J, Selzman CH, Drakos SG, McKellar SH, Wever-Pinzon O. Outcomes in Patients With Hypertrophic Cardiomyopathy Awaiting Heart Transplantation. Circ Heart Fail. 2018;11:e004378.
Lee MS, Zimmer R, Kobashigawa J. Long-Term Outcomes of Orthotopic Heart Transplantation for Hypertrophic Cardiomyopathy. Transplant Proc. 2014;46(5):1502–2150.
Mazur M, Bhat G, Popjes E, Dowling R, Eisen HJ. Long-term post-transplantation outcomes in patients with hypertrophic cardiomyopathy: Single-center 35-year experience. Clin Transplant. 2024;38(2):e15265.
Kato TS, Takayama H, Yoshizawa S, et al. Cardiac transplantation in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(4):568–74.
Biagini E, Spirito P, Leone O, et al. Heart transplantation in hypertrophic cardiomyopathy. Am J Cardiol. 2008;101(3):387–92.
Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3(5):574–9.
Rowin EJ, Maron BJ, Abt P, Kiernan MS, Vest A, Costantino F, et al. Impact of advanced therapies for improving survival to heart transplant in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2018;121:986–96.
Mazur M, Carmona Rubio A, Eisen HJ, Bhat G, Dowling R. Impact of the New Heart Allocation System on the Medium-Term Outcomes in Patients With Hypertrophic Cardiomyopathy. ASAIO J. 2024. https://doi.org/10.1097/MAT.0000000000002216.
Loyaga-Rendon RY, Fermin D, Jani M, et al. Changes in heart transplant waitlist and posttransplant outcomes in patients with restrictive and hypertrophic cardiomyopathy with the new heart transplant allocation system. Am J Transplant. 2021;21:1255–62.
Fowler CC, Helmers MR, Smood B, et al. The modified US heart allocation system improves transplant rates and decreases status upgrade utilization for patients with hypertrophic cardiomyopathy. J Heart Lung Transplant. 2021;40:1181–90.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
M.M. wrote the main manuscript and prepared the figures. M.M., E.P., W.B. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
No animal or human subjects by the authors were used in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mazur, M., Braksator, W. & Popjes, E. Hypertrophic Cardiomyopathy: From Medical Treatment to Advanced Heart Failure Therapies. Curr Cardiol Rep 26, 985–994 (2024). https://doi.org/10.1007/s11886-024-02095-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-024-02095-6