
Abstract
Purpose of Review
Increases in the availability of genetic data and advances in the tools and methods for their analyses have enabled well-powered genetic association studies that have significantly enhanced our understanding of the genetic factors underlying both rare and common valve diseases. Valvular heart diseases, such as congenital valve malformations and degenerative valve lesions, increase the risk of heart failure, arrhythmias, and sudden death. In this review, we provide an updated overview of our current understanding of the genetic mechanisms underlying valvular heart diseases. With a focus on discoveries from the past 5 years, we describe recent insights into genetic risk and underlying biological pathways.
Recent Findings
Recently acquired knowledge around valvular heart disease genetics has provided important insights into novel mechanisms related to disease pathogenesis. Newly identified risk loci associated valvular heart disease mainly regulate the composition of the extracellular matrix, accelerate the endothelial-to-mesenchymal transition, contribute to cilia formation processes, and play roles in lipid metabolism.
Summary
Large-scale genomic analyses have identified numerous risk loci, genes, and biological pathways associated with degenerative valve disease and congenital valve malformations. Shared risk genes suggest common mechanistic pathways for various valve pathologies. More recent studies have combined cardiac magnetic resonance imaging and machine learning to offer a novel approach for exploring genotype-phenotype relationships regarding valve disease. Progress in the field holds promise for targeted prevention, particularly through the application of polygenic risk scores, and innovative therapies based on the biological mechanisms for predominant forms of valvular heart diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart valves, namely the mitral valve (MV), the tricuspid valve (TV), the aortic valve (AV), and the pulmonary valve (PV), are crucial for cardiac function. Severe valvular insufficiency or stenosis is a major cause of syncope, heart failure, arrhythmias, or sudden death. Valvular heart diseases (VHD) have a reported prevalence of 2.5% in the USA [1]. While cardiac surgery can prevent major cardiac complications [2, 3], a complete understanding of VHD pathogenesis remains elusive, hindering the development of effective prevention strategies and less invasive treatment options.
The main causes of VHD are typically divided into six categories: degenerative valve disease (DVD), rheumatic heart disease (RHD), secondary valve disease, congenital malformations of the valves, valve replacement and miscellaneous [4]. Of these, VHD due to RHD, secondary valve disease, and valve replacement have limited genetic contributions. The most common congenital malformation is bicuspid aortic valve disease (BAV), with an estimated familial heritability of up to 89% [5]. DVD encompasses degenerative, calcified, fibrotic and mucinous lesions, primarily related to aging. Nonetheless, genetic factors significantly influence DVD, which covers all valvular stenosis or insufficiency, including aortic stenosis (AS), mitral valve insufficiency, and tricuspid valve insufficiency [6]. All three forms of VHD mostly follow a complex model of inheritance, involving numerous genetic risk alleles, and are influenced by external modifiers (e.g., unhealthy lifestyle for AS, mechanical stress for mitral valve prolapse (MVP)). Evidence for familial recurrence is also reported in the case of MVP [7] and calcific aortic valve stenosis (CAVS) [8]. Recent estimates from Swedish nationwide registers indicate that atrioventricular valve disease accounts for the majority of VHD cases, with AS in 47.2% of cases, mitral regurgitation (MR) in 24.2%, and aortic regurgitation in 18%. Pulmonary valve stenosis is more common in neonates, while 69% of VHD were generally diagnosed in age groups above 65 years [9].
A better understanding of the genetic and underlying biological mechanisms of VHD is critical to improving their prevention and treatment. Recently, an increasing number of studies have identified new risk genes associated with VHD and revealed novel biological pathways relevant to disease onset. This review focuses on genetic studies made available in the past 5 years, shedding light on the genetic basis of VHD. Some studies use family designs in both large or small pedigrees. Additionally, several new genome-wide associations (GWAS) have been conducted, applying statistical tests to assess the association between millions of common genetic variants (frequency > 0.01) and VHD using a case-control population-based design. GWAS have also enabled the calculation of polygenic risk scores (PRS), a promising tool for patient risk stratification.
In this review, we have summarized recent methods and results of major population-based studies focusing on the genetics of VHD. Genes identified from these studies are summarized in Table 1. In addition, we have outlined current gaps in knowledge around the genetics and multi-omics of VHD, pointing to areas that warrant future investigation.
Degenerative Lesions
Valve Calcification and Fibrosis
The causes of valve calcification are complex, involving biological mechanisms such as oxidized lipid accumulation, inflammation, myofibrillogenesis, osteogenesis, endothelial extracellular matrix (ECM) disorders, mechanobiology, and cellular senescence. Evidence suggests interactions among these mechanisms [10•]. Valvular calcification primarily affects the aortic valve and mitral annulus. However, there has been limited progress in genetic studies related to mitral annular calcification (MAC) over the past 5 years.
Calcified aortic valve disease is common in the elderly and is characterized by histological changes in the aortic valve, including ECM remodeling, osteochondral differentiation, and calcification. These changes interact to lead to aortic sclerosis, AS, and subsequently heart failure. Aortic calcification mainly manifests by aortic valve leaflet thickening, fibrosis, and mineralization of the aortic valve leaflets [11••]. Risk factors for valvular calcification include hypertension, plasma obesity, and diabetes. Importantly, age and diastolic aortic valve malformation are reported as important risk factors for CAVS [11••].
Aortic Valve Calcification and Stenosis
Several studies have shown the complexity of the genetic basis of valvular calcification, indicating an important genetic component. A study examining the surgical population in France observed familial aggregation of CAVS [8]. In a more recent and large-scale epidemiological investigation involving over 6 million Swedish siblings, among whom 13,442 had AS, an increased hazard ratio of 3.4 was observed when at least 1 sibling was affected, rising to a hazard ratio of 32 when more than 1 sibling was affected [12]. Another study based on a large pedigree cohort (2,371 relatives from 138 CAVS families) estimated the important and significant heritability of CAVS (h2 = 0.47, p < 0.0001) [13]. These findings support genetic factors to be important risk factors, potentially as prominent as the aforementioned environmental factors, likely under a complex genetic model involving a large number of common variants (Fig. 1).

Main genetic features of aortic and mitral valve major diseases. The heredity characteristics of the three most common valvular diseases are shown, including BAV, CAVD, and MVD. BAV bicuspid aortic valve, CAVD calcific aortic valve disease, CAD coronary artery disease, MVD mitral valve disease, MR mitral regurgitation, MVP mitral valve prolapse
The initial GWAS aimed at identifying common genetic variants associated with CAVS revealed a major locus near the LPA gene, supporting a genetic link between lipoproteins and CAVS. Specifically, the variant rs10455872 on LPA showed a strong association with aortic valve calcification[14]. Lp(a) is known to promote the release of calcified extracellular vesicles from primary human smooth muscle cells and valvular interstitial cells (VICs) and to increase the formation of cardiovascular calcification [15]. The established association between low-density lipoprotein cholesterol (LDL-C) levels and aortic valve stenosis risk was recently further supported using Mendelian randomization analyses, a method using naturally occurring genetic variation to investigate causal relationships in observational studies regarding how modifiable exposures affect disease [16, 17]. Moreover, recent genetic evidence supports a higher expression of the proprotein convertase subtilisin/kexin type 9 gene (PCSK9), which is correlated with elevated LDL-C levels, being associated with an increased risk of CAVS. Initially, the PCSK9 coding variant R46L, a loss-of-function mutation with a frequency of 3%, was linked to lower levels of Lp(a) and LDL cholesterol, as well as reduced risk of AS and myocardial infarction [18]. To enhance analytical power and identify additional disease-associated variants, a recent meta-analysis of ten association studies further supported the notion that mutation carriers have a lower prevalence of CAVS (odds ratio = 0.8 [0.7–0.91], p = 0.0011) [19]. PSCK9 expression levels were reported to be higher in valves from CAVS patients compared to control aortic valves, increased in VICs under a pro-osteogenic cell culture medium, and significantly decreased after treatment with a PSCK9 neutralizing antibody in vitro [18, 19].
Recent GWAS have reported novel risk variants associated with CAVS. The palmdelphin gene (PALMD) and testis expressed 41 gene (TEX41) were identified as risk factors for AS in a large dataset, including an Icelandic case-control study, and several validation GWAS with European ancestry, including the UK Biobank [20]. This study also comfirmed the association of the LPA locus with AS and revealed a rare coding variant in the myosin heavy chain 6 gene (MHY6) that increased the risk of AS, partially through its effect on BAV risk [20]. Moreover, this study utilized chromatin interaction maps from heart and aorta tissue samples to demonstrate the remote regulatory potential of associated variants on cardiac development-related promoters in the PALMD and TEX41 loci, with the difficulty of narrowing down the list of candidate genes [20]. Nonetheless, gene expression data supports the role of PALMD in AS. A transcriptome-wide association study (TWAS), a method combining GWAS involving 1009 CAVS cases and 1017 controls and expression levels of aortic valve tissues from 233 patients, revealed that lower mRNA expression of PALMD is associated with an increased risk and disease severity of CAVS [21]. In a more recent GWAS involving 14,451 CAVS patients and 398,544 controls in the Million Veteran Program, 14 significant variants were described, and these findings were replicated in several cohorts, totaling 12,889 cases and 348,094 controls [22••]. Common variants were located in 11 unique genomic regions, including 5 previously reported (PALMD, TEX41, IL6, LPA, FADS) and 6 new loci (CEP85L, FTO, SLMAP, CELSR2, MECOM, CDAN1) [22••]. Notably, the study was the first to incorporate multi-ancestry GWAS results and successfully replicated two of the loci in Hispanic (CELSR2) and African (SLMAP) ancestry case-control studies. This work contributes to improving the currently scarce genetic data available for non-European ancestry cohorts with VHD, emphasizing the need for further efforts to improve risk prediction based on genetic factors across all ancestral populations [22••]. Interestingly, this study also employed Mendelian randomization to provide evidence for the involvement of LDL cholesterol levels and obesity in CAVS via FTO, a well-established obesity risk locus.
In an in vitro calcification model using human valvular interstitial cells (hVICs) from CAVS patients, PALMD was found to be upregulated in calcified hVICs, suggesting PALMD may play a role in promoting CAVS by regulating glycolysis and NF-κB-mediated inflammation [23]. However, it remains unclear whether the causal and functional variants were equally covered in the larger GWAS [20] and TWAS [21], emphasizing the need for further investigations to formally support the role of this gene in AS. Furthermore, evidence provided from the network and pathway-enrichment analyses conducted on pre-existing transcriptomic datasets obtained from calcified and normal aortic valves have provided promising leads in aortic valve calcification [24]. This study highlighted enriched pathways related to leucocyte migration and ECM constituents. Additionally, it proposed SPP1, TNC, SCG2, FAM20A, and CD52 as hub genes of the top 5 enriched networks using weighted gene co-expression networks [24]. Network analyses are valuable for identifying biological mechanisms and assessing the probabilistic relevance of changes in gene expression for a specific condition. Nonetheless, further validation in larger and independent datasets backed with further experimental evidence is warranted to confirm the role of these hub genes in the pathogenesis of aortic calcification.
Genetic studies on CAVS have highlighted impaired metabolism as an additional causal mechanism. In a large GWAS involving ~ 9400 CAVS cases and ~ 300,000 controls, common variants within the fatty acid desaturase 1 and 2 genes (FADS1/2) locus were associated with a higher risk of AS and CAVS [25•]. FADS1/2 are involved in ω-6 and ω-3 fatty acid biosynthesis, and this genetic locus is known to influence LDL cholesterol and triglycerides levels, in addition to asthma and coronary artery disease [25•]. Increased expression of arachidonic acid, a product of FADS1 in the liver, was associated with AS, along with genetically elevated levels of ω-6 and ω-3 fatty acids [25•]. Interestingly, a recent investigation of untargeted global metabolomic data obtained from valve tissue of 96 human donors with varying degrees of CAVS severity identified lipid metabolism and biosynthesis (glycerophospholipid metabolism), linoleic acid metabolism, and bile acid biosynthesis as the top 3 significantly altered metabolomic pathways associated with CAVS severity[26].
Mitral Annulus Calcification
The mitral annulus is a fibrous structure surrounding the mitral valve leaflets that is susceptible to calcification, with a prevalence estimated to range from 5 to 42%, frequently observed in older subjects [27]. MAC is commonly associated with hypertrophic cardiomyopathy, as estimated in a study that reported MAC cases in 18.5% of these patients [28]. Furthermore, MAC has also been linked to chronic kidney disease and hypertension [27]. However, the pathophysiology and genetic mechanisms of mitral annular calcification have not been investigated in-depth, apart from a GWAS involving 3795 participants, which yielded inconclusive results regarding 2 common variants in the interleukin 36 gamma gene (IL36G, alias IL1F9) [14].
In summary, recent data confirm that the etiological mechanisms of CAVS are intricate. CAVS exhibits genetic associations not only with cardiac developmental processes, the extracellular matrix, valvular endothelial cells, and mesenchymal cells but also with lipoprotein levels and metabolism. On the other hand, regarding MAC, current findings have suggested that its pathogenesis likely shares similarities with atherosclerosis and CAVS, although dedicated investigations focusing on MAC to elucidate its specific genetic causes are still lacking.
Degenerative Valve Diseases
Mitral Valve Disease
Calcification of the mitral valve mostly occurs in the mitral annulus, whereas calcification of the valve leaflets is less common. Mitral valve prolapse (MVP) and mitral regurgitation (MR) are the main lesions associated with the mitral valve. In the USA, the prevalence of MR is approximately 1.7%, but it increases to up to 9.3% in individuals > 75 years old [29]. MVP, with a prevalence of about 2.4%, is a common cause of primary MR in developed countries and is categorized into two major phenotypes: a mucinous malformation associated with redundancy of the anterior and posterior mitral leaflets and tendon cords, identified as Barlow disease when detected early in life, and elastin deficiency (FED), a more degenerative form that is particularly prevalent in older individuals (Fig. 1). However, it remains uncertain whether those two forms of MVP represent different diseases or distinct manifestations of the same disease [30]. MVP is currently under active genetic investigation, with important progress in understanding its genetic causes over the past 5 years, encompassing both familial studies and GWAS. As for now, no specific genetic causes have been described for FED, and current studies have treated both MVP phenotypes as a single entity. The same approach applies to MR.
Familial Mitral Valve Prolapse
The filamin A gene (FLNA) was the first gene discovered through genetic linkage analysis in a large French family to be caused by isolated myxomatous polyvalvular dystrophic and nonsyndromic MV disease (MVD) [31, 32]. FLNA is a cytoskeletal protein that plays a role in regulating cell shape and migration [33]. In a recent clinical study, genotype-to-phenotype correlations were assessed in 246 subjects, including 72 mutation carriers from four FLNA-MVD families with three different FLNA mutations [34]. The study revealed that MVP caused by FLNA mutations exhibits a specific phenotype, with restricted leaflet motion in diastole and papillary muscles positioned closer to the mitral annulus. These findings support the notion of a separated phenotype from classical Barlow disease or FED [34]. Of note, genetic variations identified in FLNA so far are extremely rare and may be potentially specific to the genetic isolates in this particular region.
A recent study, by Toomer et al. [35] employed a combination of human pedigree analysis, mouse models, and population genetics to explore the role of primary cilia in the etiology of MVP. The study investigated the role of several genes involved in cilia structure and biology, along with their expression in VICs. The study also shed light on the involvement of cilia in ECM deposition during cardiac and valve development by employing knockout mice models of genes encoding major cilium components [35]. The authors first performed genetic screening for coding and relevant mutations in informative family members. They employed targeted sequencing of a genomic region previously linked significantly to MVP. After applying various filters based on variant frequency, potential biological significance concerning the position of the genetic variants in coding sequences of candidate genes, and segregation in families with MVP, they followed up a missense mutation in the cilia gene DAZ interacting zinc finger protein 1 (DZIP1) [35]. Subsequently, a new mouse model for MVP carrying this point mutation knock-in Dzip1 was generated, which led to cilia loss. This, in turn, resulted in dysregulation of ECM synthesis during valve development and myxomatous mitral valve in adult mice [35]. Moreover, the study assessed a set of 278 cilia-related genes for their association with MVP using existing case-control GWAS data. The results revealed a significant enrichment for MVP-associated variants within this specific group of genes, providing further support for the potential role of this mechanism in the genetic susceptibility to sporadic MVP [35]. However, translating ciliary defects during cardiac development into potential therapeutic strategies for MVP poses a challenge and needs further investigation.
Sporadic Mitral Valve Prolapse
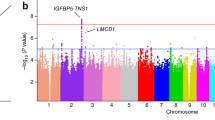
In recent years, multiple efforts have been made to enhance our understanding of the genetic basis of MVP using GWAS-based strategies. The first GWAS on sporadic MVP identified 6 association signals, including those near tensin 1 (TNS1) on chromosome 2, and LIM and cysteine-rich domains 1 (LMCD1) on chromosome 3 [36]. Knockdown of both genes in zebrafish showed atrioventricular valve regurgitation, while enlarged posterior mitral leaflets were observed in Tns−/− mice [36]. Furthermore, the recent availability of mitral valve genome-wide open chromatin profiles, a genomic map, which identify genomic regions likely involved in gene expression regulation in the mitral valve, has facilitated functional annotation of MVP risk loci [37•]. This has helped narrow down the list of causal variants in some of the previously identified loci, which is an essential step in the selection of GWAS risk loci to gain a better understanding of how non-coding variants may influence the gene expression of nearby candidate genes [37•]. Functional insights were complemented by reporter assays, enhancer deletions in human fibroblasts, and circular chromatin conformation capture techniques, elucidated the regulation role of a long-range enhancer for the TNS1 gene [37•]. It was also revealed that regulatory features of nuclei from normal and myxomatous mitral valve tissues substantially differ from those of myocardium tissue and cardiac fibroblasts, with a significant enrichment for MVP-associated variants specifically observed in open chromatin maps from mitral valve tissues [37•]. In addition, leveraging the initial MVP GWAS dataset, an additional locus in the GLIS family zinc finger 1 gene (GLIS1) was identified through pathway-enrichment analyses [38]. These analyses combined the MVP-associated variants from previously reported GWAS with specific pre-established pathways based on gene functions (e.g., KEGG database, gene-ontology). This approach offers the advantage of increasing the power of genetic association analyses by analyzing groups of genes instead of individual variants. As a result, this analysis substantiated GLIS1 as an MVP risk locus by the inclusion of a case-control dataset from the UKB [38]. GLIS1 encodes a transcription factor from the Krüppel-like zinc finger family associated with the EMT process during valve degeneration [38]. Overall, these findings further support that the mechanisms involved in cardiac development are particularly enriched through pathway analyses of genetic risk variants associated with both sporadic and more common forms of MVP [38].
A recent genome-wide association meta-analysis of MVP involved 4,884 cases and 434,649 controls, making it the largest genetic study of MVP to date [39••]. This study was supported by extensive multi-omics functional annotations, including the myxomatous and non-myxomatous open chromatin datasets [37•]. The study identified 14 MVP-associated risk loci and 33 candidate genes, including previously identified genes like TNS1 and LMCD1, as well as newly discovered genes such as latent transforming growth factor beta binding protein 2 gene (LTBP2) and spectrin beta, non-erythrocytic 1 (SPTBN1) [39••]. Interestingly, this study showed that individuals in the top 20% of a polygenic risk score generated from the MVP GWAS results had a 1.79-fold increased risk of MVP in comparison to the remaining 80% of individuals [39••]. This initial glimpse into the potential use of polygenic risk scores for MVP suggests their future value in clinical applications as a practical, cost-effective tool for predicting patient risk. However, given limitations related to sample size and the absence of validation in diverse populations, additional efforts are required to enhance the statistical reliability of MVP PRS and determine their broader applicability in the clinical setting.
Recent investigations on DVD have confirmed the existence of various phenotypes and a complex, polygenic inheritance pattern. Significant advancements have been achieved, especially using multi-omics approaches that are now available for MVP. Nevertheless, we have an incomplete understanding of the highly polygenic nature of common types of VHD. Additional factors, such as the risk of mitral regurgitation and the FED phenotype, as well as the combined risk with other cardiac diseases like arrythmias and cardiomyopathies, remain to be explored. Moreover, we also expect that the current limitations in applying PRS to MVP will be substantially addressed by ongoing efforts that involve larger and more diverse GWAS datasets.
Tricuspid Regurgitation
The incidence of tricuspid regurgitation is relatively low when compared to mitral valve and aortic valve disease, estimated at 2.6% in the USA (Fig. 1) [40]. However, detecting tricuspid regurgitation can be challenging due to the lack of obvious symptoms associated with this condition. A study has shown a large variation in tricuspid valve morphology by ultrasound testing, with about half of patients having three leaflets, while 39% have four [41]. Tian et al. investigated the role of IncRNAs in tricuspid regurgitation-induced right ventricular cardiomyopathy [42]. Despite these efforts, there remains a gap in genetic studies related to both familial and sporadic tricuspid regurgitation.
Congenital Valvular Malformations
The primary congenital valvular malformations include BAV and the less studied pulmonary valve stenosis (PVS). PVS typically occurs as isolated congenital defects in over 95% of cases and is also related to rare syndromes such as Noonan Syndrome, accounting for about 7% of congenital heart disease (Fig. 1) [43]. In children, the clinical course of PVS is usually benign with a high survival rate. However, as individuals age, the disease may be aggravated due to fibrous thickening of the valve and, in rare instances, valve calcification, driven by poorly understood biological mechanisms.
BAV has been the focus of active investigation in recent years. It has been reported as a highly heritable VHD, with a prevalence of 1% in the population. Within families, the heritability estimate (h2) for BAV reached up to 89% (Fig. 1) [5]. A recent echocardiography-based study of first-degree relatives of BAV patients demonstrated the high heritability of BAV and reported a prevalence of 6.4%, which is over three times the expected rate [44]. Studies of the genetic basis of BAV have emphasized familial inheritance. For instance, the investigation of the notch receptor 1 gene (NOTCH1), a well-established genetic cause of BAV, revealed that these mutations explained 2% of familial BAV but less than 0.1% of sporadic cases [45•]. Using WES in a three-generation family, researchers aimed to understand variants located at all protein-coding regions of genes that might underlie symptoms or disease. This study revealed the role of NOTCH1 mutations in familial heart disease and represented diverse clinical phenotypes of BAV, including mainly ventricular septal defect, aortic valve calcification, and thoracic aortic aneurysm [46].
In the last 5 years, there has been significant progress in identifying novel genetic causes of BAV through targeted sequencing, which provides a cost/time-effective approach for studying specific genes of interest. In a small-scale genotyping-based analysis of 152 unrelated BAV patients and 200 matched healthy individuals, a novel heterozygous stop mutation p.E386X in GATA binding protein 6 (GATA6) was identified [47]. This mutation led to an inactivated protein and disrupted the synergistic transcriptional activation mechanism with another important transcription factor of valve development encoded by the GATA binding protein 4 (GATA4) [47]. Likewise, a heterozygous mutation in GATA4, p.E147X, was reported to inactivate GATA4 and impair its transcriptional activation by NK2 homeobox 5 (NKX2.5) [48]. In human valves, lower GATA6 transcript levels were associated with BAV. These altered transcript levels disrupted valve remodeling function and changes in the extracellular matrix composition in valves from Gata6+/− mice [49]. These animal models also exhibited a highly permeable right-left type of BAV [49].
In addition to NOTCH1 and GATA4/6, recent studies have identified heterozygous mutations in roundabout guidance receptor 4 gene (ROBO4) in two families that presented BAV and ascending aortic aneurysm [50]. These mutations in ROBO4 were reported to impair the endothelial barrier function of human aortic endothelial cells in vitro. They also led to EMT cell conversion. In full knockout mice, complex cardiovascular phenotypes were observed, including aortic valve thickening, sometimes with BAV, stenosis, and/or ascending aorta aneurysm [50]. Additionally, nonsynonymous mutations in roundabout guidance receptor 1 gene ROBO1 were found in family with BAV and multiple valvular diseases [51]. This provides further evidence for the involvement of the ROBO gene family in BAV. Notably, members of this family also carried mutations in GATA binding protein 5 gene GATA5, representing another example of incomplete intrafamilial penetrance of BAV and a complex inheritance pattern [51].
Through WGS of the coding and non-coding regions of DNA in five small nuclear families, the cadherin EGF LAG seven-pass G-type receptor 1 (CELSR1) was proposed as a potential risk gene for familial BAV associated with hypoplastic left heart syndrome [52]. This study also identified mutations in CELSR1 as compound heterozygous with mutations in CELSR3 and myosin XVA gene (MYO15A) [52]. Under the hypothesis of ciliogenesis as a potential mechanism for valve diseases, supported by zebrafish and mouse models, a GWAS involving 452 BAV cases and 1834 controls reported several exocyst complex component genes (EXOC4, EXOC6, EXOC8, and EXOC5) as potential genetic causes of BAV, valvular calcification, and stenosis [53]. Similarly, another GWAS found that mucin 4 gene (MUC4), a cell surface-associated gene, plays an important role in the EMT process associated with BAV [54]. Knocking out Muc4 in zebrafish resulted in delayed cardiac development and potential aortic valve malformation [54]. Importantly, these more recent BAV GWAS also reported significant associated variants in PALMD and GATA4 loci, which had been previously reported [20, 54, 55].
Interestingly, while impaired metabolism is a significant factor in CAVS, a study comparing tricuspid aortic valve patients to BAV patients revealed that those with BAV exhibited an upregulation of arachidonic acid metabolism and a decrease in arginine and proline metabolites following valve replacement. These findings suggest that multiple metabolic dysfunctions, including elevated oxidative stress, inflammation, and impaired NO production, may be related to worse left ventricular reverse remodeling after tricuspid aortic valve repair in BAV [56]. This insight sheds light on the complex metabolic aspects of BAV and its implications for surgical outcomes.
In summary, recent studies have reported genetic mutations, both within established genes like NOTCH1 and in newly identified ones, mostly involved in alteration of the extracellular matrix composition, acceleration of the EMT transformation, or ciliogenesis as causal mechanisms underlying BAV and its calcification. These studies based on a combination of family pedigrees and population studies have provided valuable insights into BAV. However, these findings are insufficient to elucidate the complete pathogenesis of this common VHD, such as the mechanisms associated with calcification that affects both BAV and tricuspid aortic valve (TAV) that still need to be fully determined.
Research Driven from Structural Phenotypes of the Heart
In addition to genetic factors associated with VHD, there is a growing body of evidence that abnormal blood stress can contribute to pathological changes in heart valves [57]. However, the biological mechanisms underlying the mechanical stress that leads to structural changes in the valves, potentially culminating in valvular diseases, remain inadequately understood. In addition to mechanical stress, factors such as aging, inflammation, and calcification have been associated with valve diseases. These conditions are directly characterized by structural changes in the valve leaflets and annulus, which can be identified by imaging techniques. Therefore, in addition to the traditional case-control study of valvular diseases, the study of phenotypes related to valvular structure is now contributing to our understanding of the pathogenesis of VHD.
In recent years, the advancement of machine learning has facilitated the automated extraction of human heart phenotypes from cardiac magnetic resonance (CMR) images, encompassing structural, functional, tissue characteristics, hemodynamics, and metabolic measurements [58]. Analyzing the genetic markers associated with these phenotypes and investigating their relationships with non-image phenotypes and diseases has become an important approach for studying the complex pathogenic mechanisms of cardiovascular disease, including VHDs. Córdova et al. investigated the genetic mechanisms of the aortic valve area, which is estimated by planimetry from CMR imaging sequences of the aortic valve from UK Biobank (UKB). Through GWAS, they identified three significant loci (deleted in lymphocytic leukemia 1 (DLEU1), CASP2 And RIPK1 domain containing adaptor with death domain (CRADD), and Golgi SNAP receptor complex member 2 (GOSR2)) associated with multiple cardiovascular diseases [59]. PRS analyses indicated that a smaller aortic valve area might predict an increased risk of clinically defined aortic valve disease (odds ratio = 1.14; p = 2.3 × 10−6). The same research group also investigated the genetic mechanisms of mitral annular diameter, a critical criterion for valvular function. They performed automated measurements of the mitral valve annular diameter in the 4-chamber view using 32,220 MRI images from the UK Biobank at ventricular systole and diastole, which served as the phenotypes for their GWAS analyses. In this study, they identified ten risk loci, four of which were located at four new genes (GOSR2, Erb-B2 receptor tyrosine kinase 4 (ERBB4), multiple C2 and transmembrane domain containing 2 (MCTP2), microcephalin 1 (MCPH1)) related to the formation, growth, and functional maintenance of cardiac valves. The study also found three known cardiac contractility-related genes (BAG Cochaperone 3 (BAG3), Titin (TTN), RNA Binding Fox-1 Homolog 1(RBFOX1)), which did not overlap with the genetic risks of MAC [14]. By integrating the findings with ATAC-Seq data from primary mitral valve tissue [37•], rs17608766 was localized in open chromatin regions, suggesting its role as a potential enhancer element in GOSR2, a gene associated with both aortic and mitral valve biology. Of note, the PRS analysis revealed that polygenic scores for systolic mitral annular diameter were associated with the risk of MVP (odds ratio = 1.19; p = 4.9 × 10−11) [60•].
Even though it is promising to study VHDs using imaging-based phenotypes, there are limitations to this approach. These limitations include challenges related to the imaging quality, the accuracy of the algorithm used to translate images into phenotypes, and the capacity to effectively integrate various disease-related phenotypes. Despite these challenges, the above studies serve as proof of concept, demonstrating the potential to use quantitative imaging phenotypes to accelerate the discovery of genes and mechanisms associated with VHDs.
Conclusion
Currently, most studies on the genetics of VHD have focused on the aortic and mitral valves, which represent the most common types of VHD. The field of genetics has seen substantial growth, offering mounting evidence for a genetic contribution to various VHDs. VHDs may be congenital, or caused by rare, highly-penetrant mutations affecting several cardiac functions, and manifest as rare syndromes, as sometimes observed with pulmonary stenosis. Even in the case of a shared genetic model, some VHDs like BAV demonstrate high heritability and familial recurrence, while CAVS that affect the same heart valve display an important polygenic contribution and complex genetic architecture. In the case of CAVS, lifestyle and environmental causes play a significant role, along with additional cardiovascular comorbidities including coronary artery disease, hypertension, and dyslipidemia. On the other hand, we displayed several examples where genetic and biological mechanisms are shared between VHDs, even those affecting different valves. For example, BAV and myxomatous MVP share genes like SPTBN1, which is involved in ciliogenesis as a common mechanism [20, 23, 39, 52, 54]. One of the future challenges will be to define and explore the similarities and differences in genetic mechanisms between distinct VHDs. Evidence from single-cell sequencing explorations indicate important heterogeneity in cell types within valve tissue. It is likely that risk genes involved in heart valve pathogenesis act through different cell subpopulations for different valve diseases. The recent application of deep learning to leverage cardiac MRI phenotypes within large-scale datasets has provided innovative methodologies for assessing the genetics of VHDs. One significant limitation in this field is the absence of specific expression data in most valves to generate well-powered genetically determined gene expression datasets, similar to what has been available for the mitral valve. Despite inherent challenges with accessing heart valve tissue, efforts are needed to provide powerful multi-omics resources to complement the impressive progress made through GWAS in recent years. Finally, we are still lacking well-powered multi-ethnic and sex-stratified studies despite encouraging recent initiatives [22••]. Altogether, we expect significant advances in VHD genetics in the coming years, which will be crucial to comprehensively explore the diverse mechanisms underlying VHD and establish a solid scientific foundation to improve the current tools for prediction and diagnosis. Moreover, these developments may lead to promising, alternative treatments, which may reduce the current reliance on surgical and interventional management of VHD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127(1):143–52. https://doi.org/10.1161/CIR.0B013E318282AB8F.
Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368(9540):1005–1011. https://doi.org/10.1016/S0140-6736(06)69208-8.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139-e596. https://doi.org/10.1161/CIR.0000000000000757.
Yang Y, Wang Z, Chen Z, Wang X, Zhang L, Li S, et al. Current status and etiology of valvular heart disease in China: a population-based survey. BMC Cardiovasc Disord. 2021;21(1):339. https://doi.org/10.1186/S12872-021-02154-8.
Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson D. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44(1):138–43. https://doi.org/10.1016/J.JACC.2004.03.050.
Kodali SK, Velagapudi P, Hahn RT, Abbott D, Leon MB. Valvular heart disease in patients ≥80 years of age. J Am Coll Cardiol. 2018;71(18):2058-2072. https://doi.org/10.1016/J.JACC.2018.03.459.
Delling FN, Rong J, Larson MG, Lehman B, Osypiuk E, Stantchev P, et al. Familial clustering of mitral valve prolapse in the community. Circulation. 2015;131(3):263-268. https://doi.org/10.1161/CIRCULATIONAHA.114.012594.
Probst V, Le Scouarnec S, Legendre A, Jousseaume V, Jaafar P, Nguyen JM, et al. Familial aggregation of calcific aortic valve stenosis in the western part of France. Circulation. 2006;113(6):856-860. https://doi.org/10.1161/CIRCULATIONAHA.105.569467.
Andell P, Li X, Martinsson A, Andersson C, Stagmo M, Zöller B, et al. Epidemiology of valvular heart disease in a Swedish nationwide hospital-based register study. Heart. 2017;103(21):1696-1703. https://doi.org/10.1136/HEARTJNL-2016-310894.
• Blaser MC, Kraler S, Lüscher TF, Aikawa E. Network-guided multiomic mapping of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2023;43(3):417–26. https://doi.org/10.1161/ATVBAHA.122.318334. In this review, the authors cite recent efforts to apply epigenomics, transcriptomics, proteomic, and metabolic initiatives to study aortic valve calcification.
•• Moncla L-HM, Briend M, Bossé Y, Mathieu P. Calcific aortic valve disease: mechanisms, prevention and treatment. Nat Rev Cardiol. 2023;20(8):546–59. https://doi.org/10.1038/s41569-023-00845-7. This review provides a comprehensive summary of the risk factors, genetics, and molecular mechanisms involved in CAVD.
Martinsson A, Li X, Zöller B, Andell P, Andersson C, Sundquist K, et al. Familial aggregation of aortic valvular stenosis: a nationwide study of sibling risk. Circ Cardiovasc Genet. 2017;10(6):e001742. https://doi.org/10.1161/CIRCGENETICS.117.001742/-/DC1
Boureau AS, Karakachoff M, Le Scouarnec S, Capoulade R, Cueff C, de Decker L, et al. Heritability of aortic valve stenosis and bicuspid enrichment in families with aortic valve stenosis. Int J Cardiol. 2022;359:91–8. https://doi.org/10.1016/j.ijcard.2022.04.022.
Thanassoulis G, Campbell CY, Owens D, Smith J, Smith AV, Peloso G, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–12. https://doi.org/10.1056/NEJMoa1109034.
Rogers MA, Atkins SK, Zheng KH, Singh SA, Chelvanambi S, Pham TH, et al. Lipoprotein(a) induces vesicular cardiovascular calcification revealed with single-extracellular vesicle analysis. Front Cardiovasc Med. 2022;9:778919. https://doi.org/10.3389/FCVM.2022.778919/BIBTEX.
Nazarzadeh M, Pinho-Gomes A-C, Bidel Z, Dehghan A, Canoy D, Hassaine A, et al. Plasma lipids and risk of aortic valve stenosis: a Mendelian randomization study. Eur Heart J. 2020;41:3913–20. https://doi.org/10.1093/eurheartj/ehaa070. This Mendelian randomization-based study provided robust support for the causal association link between plasma lipids and AVS.
Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2(1):6. https://doi.org/10.1038/s43586-021-00092-5.
Langsted A, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Kamstrup PR. PCSK9 R46L loss-of-function mutation reduces lipoprotein(a), LDL cholesterol, and risk of aortic valve stenosis. J Clin Endocrinol Metab. 2016;101(9):3281–7. https://doi.org/10.1210/JC.2016-1206.
Perrot N, Valerio V, Moschetta D, Boekholdt SM, Dina C, Chen HY, et al. Genetic and in vitro inhibition of PCSK9 and calcific aortic valve stenosis. JACC Basic Transl Sci. 2020;5(7):649–61. https://doi.org/10.1016/j.jacbts.2020.05.004.
Helgadottir A, Thorleifsson G, Gretarsdottir S, Stefansson OA, Tragante V, Thorolfsdottir RB, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun. 2018;9(1):987. https://doi.org/10.1038/s41467-018-03252-6.
Thériault S, Gaudreault N, Lamontagne M, Rosa M, Boulanger MC, Messika-Zeitoun D, et al. A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat Commun. 2018;9(1):988. https://doi.org/10.1038/s41467-018-03260-6.
•• Small AM, Peloso G, Linefsky J, Aragam J, Galloway A, Tanukonda V, et al. Multiancestry genome-wide association study of aortic stenosis identifies multiple novel loci in the million veteran program. Circulation. 2023;147(12):942–55. https://doi.org/10.1161/circulationaha.122.061451. This study is one of the largest and most ancestrally diverse GWAS of AS, which identified 14 risk loci located in 11 unique genomic regions including 5 previously reported loci and 6 new loci. Two of the loci have been replicated in Hispanic and Black individuals.
Wang S, Yu H, Gao J, Chen J, He P, Zhong H, et al. PALMD regulates aortic valve calcification via altered glycolysis and NF-κB-mediated inflammation. J Biol Chem. 2022;298(5):101887. https://doi.org/10.1016/j.jbc.2022.101887.
Sun JY, Hua Y, Shen H, Qu Q, Kan JY, Kong XQ, et al. Identification of key genes in calcific aortic valve disease via weighted gene co-expression network analysis. BMC Med Genomics. 2021;14(1):135. https://doi.org/10.1186/S12920-021-00989-W/FIGURES/6.
Chen HY, Cairns BJ, Small AM, Burr HA, Ambikkumar A, Martinsson A, et al. Association of FADS1/2 locus variants and polyunsaturated fatty acids with aortic stenosis. JAMA Cardiol. 2020;5(6):694–702. https://doi.org/10.1001/JAMACARDIO.2020.0246. This GWAS provided the first evidence for the genetic role of FADS1/2 involved in ω-6 and ω-3 fatty acid biosynthesis in the risk of AS and calcification.
Surendran A, Edel A, Chandran M, Bogaert P, Hassan-Tash P, Asokan AK, et al. Metabolomic signature of human aortic valve stenosis. JACC Basic to translational science. 2020;5(12):1163–77. https://doi.org/10.1016/j.jacbts.2020.10.001.
Massera D, Kizer JR, Dweck MR. Mechanisms of mitral annular calcification. Trends Cardiovasc Med. 2020;30(5):289–95. https://doi.org/10.1016/J.TCM.2019.07.011.
Patlolla SH, Schaff HV, Nishimura RA, Geske JB, Lahr BD, Lee AT, et al. Mitral annular calcification in obstructive hypertrophic cardiomyopathy: prevalence and outcomes. Ann Thorac Surg. 2022;114(5):1679–87. https://doi.org/10.1016/j.athoracsur.2021.09.077.
Kataria R, Castagna F, Madan S, Kim P, Saeed O, Adjepong YA, et al. Severity of functional mitral regurgitation on admission for acute decompensated heart failure predicts long-term risk of rehospitalization and death. J Am Heart Assoc. 2022;11(1):e022908. https://doi.org/10.1161/JAHA.121.022908.
Flint N, Raschpichler M, Rader F, Shmueli H, Siegel RJ. Asymptomatic degenerative mitral regurgitation: a review. JAMA Cardiol. 2020;5(3):346-355. https://doi.org/10.1001/JAMACARDIO.2019.5466.
Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115(1):40–49. https://doi.org/10.1161/CIRCULATIONAHA.106.622621.
Kyndt F, Schott JJ, Trochu JN, Baranger F, Herbert O, Scott V, et al. Mapping of X-linked myxomatous valvular dystrophy to chromosome Xq28. Am J Hum Genet. 1998;62(3):627–632. https://doi.org/10.1086/301747.
Deng W, Lopez-Camacho C, Tang JY, Mendoza-Villanueva D, Maya-Mendoza A, Jackson DA, et al. Cytoskeletal protein filamin A is a nucleolar protein that suppresses ribosomal RNA gene transcription. Proc Natl Acad Sci U S A. 2012;109(5):1524-1529. https://doi.org/10.1073/PNAS.1107879109/-/DCSUPPLEMENTAL/PNAS.201107879SI.PDF.
Le Tourneau T, Le Scouarnec S, Cueff C, Bernstein D, Aalberts JJJ, Lecointe S, et al. New insights into mitral valve dystrophy: a Filamin-A genotype–phenotype and outcome study. Eur Heart J. 2018;39(15):1269-1277. https://doi.org/10.1093/EURHEARTJ/EHX505.
Toomer KA, Yu M, Fulmer D, Moore KS, Moore R, et al. Primary cilia defects causing mitral valve prolapse. Sci Transl Med. 2019;11(493):eaax0290. https://doi.org/10.1126/SCITRANSLMED.AAX0290.
Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapsed. Nat Genet. 2015;47(10):1206-1211. https://doi.org/10.1038/ng.3383.
• Kyryachenko S, Georges A, Yu M, Barrandou T, Guo L, Bruneval P, et al. Chromatin accessibility of human mitral valves and functional assessment of MVP risk loci. Circ Res. 2021;128(5):e84–101. https://doi.org/10.1161/CIRCRESAHA.120.317581. This article described unprecedented genome-wide open chromatin profiles from human pathogenic and nonpathogenic MVs.
Yu M, Georges A, Tucker NR, Kyryachenko S, Toomer K, Schott JJ, et al. Genome-wide association study-driven gene-set analyses, genetic, and functional follow-up suggest Glis1 as a susceptibility gene for mitral valve prolapse. Circ Genom Precis Med. 2019;12(5):e002497. https://doi.org/10.1161/CIRCGEN.119.002497
•• Roselli C, Yu M, Nauffal V, Georges A, Yang Q, Love K, et al. Genome-wide association study reveals novel genetic loci: a new polygenic risk score for mitral valve prolapse. Eur Heart J. 2022;43(17):1668–80. https://doi.org/10.1093/eurheartj/ehac049. This study is the largest GWAS published for MVP to date, which identified 14 risk loci, including those previously found and new genes, as well as presented the first application of PRS assessment in MVP.
Cahill TJ, Prothero A, Wilson J, Kennedy A, Brubert J, Masters M, et al. Community prevalence, mechanisms and outcome of mitral or tricuspid regurgitation. Heart. 2021;107(12):1003-1009. https://doi.org/10.1136/HEARTJNL-2020-318482.
Hahn RT, Weckbach LT, Noack T, Hamid N, Kitamura M, Bae R, et al. Proposal for a standard echocardiographic tricuspid valve nomenclature. JACC Cardiovasc Imaging. 2021;14(7):1299–1305. https://doi.org/10.1016/J.JCMG.2021.01.012.
Tian C, Yang Y, Ke Y, Yang L, Zhong L, Wang Z, et al. Integrative analyses of genes associated with right ventricular cardiomyopathy induced by tricuspid regurgitation. Front Genet. 2021;12:708275. https://doi.org/10.3389/FGENE.2021.708275/BIBTEX.
Stephensen SS, Sigfusson G, Eiriksson H, Sverrisson JT, Torfason B, Haraldsson A, et al. Congenital cardiac malformations in Iceland from 1990 through 1999. Cardiol Young. 2004;14(4):396–401. https://doi.org/10.1017/S1047951104004081.
Galian-Gay L, Carro Hevia A, Teixido-Tura G, Rodriguez Palomares J, Gutierrez-Moreno L, Maldonado G, et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart. 2019;105(8):603-608. https://doi.org/10.1136/heartjnl-2018-313802.
• Debiec R, Hamby S, Jones P, Safwan KA, Sosin M, Hetherington S, et al. Contribution of NOTCH1 genetic variants to bicuspid aortic valve and other congenital lesions. Heart. 2022;108(14):1114–20. https://doi.org/10.1136/heartjnl-2021-320428. This study assessed NOTCH1 mutations in a large number of pedigrees and patient cohorts and estimated these genes to be causal in 2% of familial and <0.1% of sporadic non-syndromic BAV.
Debiec R, Hamby SE, Jones PD, Coolman S, Asiani M, Kharodia S, et al. Novel loss of function mutation in NOTCH1 in a family with bicuspid aortic valve, ventricular septal defect, thoracic aortic aneurysm, and aortic valve stenosis. Mol Genet Genomic Med. 2020;8(10):e1437. https://doi.org/10.1002/MGG3.1437
Xu YJ, Di RM, Qiao Q, Li XM, Huang RT, Xue S, et al. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene. 2018;663:115–120. https://doi.org/10.1016/J.GENE.2018.04.018.
Li RG, Xu YJ, Wang J, Liu XY, Yuan F, Huang RT, et al. GATA4 loss-of-function mutation and the congenitally bicuspid aortic valve. Am J Cardiol. 2018;121(4):469–474. https://doi.org/10.1016/j.amjcard.2017.11.012.
Gharibeh L, Komati H, Bossé Y, Boodhwani M, Heydarpour M, Fortier M, et al. GATA6 regulates aortic valve remodeling, and its haploinsuffciency leads to right-left type bicuspid aortic valve. Circulation. 2018;138(10):1025–1038. https://doi.org/10.1161/CIRCULATIONAHA.117.029506.
Gould RA, Aziz H, Woods CE, Seman-Senderos MA, Sparks E, Preuss C, et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. 2018;51(1):42–50. https://doi.org/10.1038/s41588-018-0265-y.
Jaouadi H, Gérard H, Théron A, Collod-Béroud G, Collart F, Avierinos JF, et al. Identification of non-synonymous variations in ROBO1 and GATA5 genes in a family with bicuspid aortic valve disease. J Hum Genet. 2022;67(9):515–518. https://doi.org/10.1038/s10038-022-01036-x.
Theis JL, Niaz T, Sundsbak RS, Fogarty ZC, Bamlet WR, Hagler DJ, et al. CELSR1 risk alleles in familial bicuspid aortic valve and hypoplastic left heart syndrome. Circ Genom Precis Med. 2022;15(2):e003523. https://doi.org/10.1161/CIRCGEN.121.003523.
Fulmer D, Toomer K, Guo L, Moore K, Glover J, Moore R, et al. Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. 2019;140(16):1331–1341. https://doi.org/10.1161/CIRCULATIONAHA.119.038376.
Gehlen J, Stundl A, Debiec R, Fontana F, Krane M, Sharipova D, et al. Elucidation of the genetic causes of bicuspid aortic valve disease. Cardiovasc Res. 2023;119(3):857–866. https://doi.org/10.1093/CVR/CVAC099.
Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M, et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun. 2017;8:15481. https://doi.org/10.1038/ncomms15481.
Xiong T-Y, Liu C, Liao Y-B, Zheng W, Li Y-J, Li X, et al. Differences in metabolic profiles between bicuspid and tricuspid aortic stenosis in the setting of transcatheter aortic valve replacement. BMC Cardiovasc Disord. 2020;20(1):229. https://doi.org/10.1186/s12872-020-01491-4.
O’Donnell A, Yutz KE. Mechanisms of heart valve development and disease. Development (Cambridge). 2020;147. https://doi.org/10.1242/DEV.183020/224244.
Wang C, Li Y, Lv J, Jin J, Hu X, Kuang X, et al. Recommendation for cardiac magnetic resonance imaging-based phenotypic study: Imaging part. Phenomics. 2021;1(14):151–170. https://doi.org/10.1007/S43657-021-00018-X.
Córdova-Palomera A, Tcheandjieu C, Fries JA, Varma P, Chen VS, Fiterau M, et al. Cardiac imaging of aortic valve area from 34 287 UK Biobank participants reveals novel genetic associations and shared genetic comorbidity with multiple disease phenotypes. Circ Genom Precis Med. 2020;13(6):e003014. https://doi.org/10.1161/CIRCGEN.120.003014.
• Yu M, Tcheandjieu C, Georges A, Xiao K, Tejeda H, Dina C, et al. Computational estimates of annular diameter reveal genetic determinants of mitral valve function and disease. JCI Insight. 2022;7(3):e146580. https://doi.org/10.1172/jci.insight.146580Based on the automated estimates of mitral valve annular diameter from MRI images from the UK Biobank, this study identified 10 risk loci, including GOSR2. The polygenic scoring of MV annular diameter in systole was predictive of risk MVP.
Funding
This work was sponsored by the Shanghai Sailing Program (21YF1452900) and Shanghai Clinical Research Program (20234Y0239) to Dr Yu. NBN is supported by the European Research Council grant (ERC-Stg-ROSALIND-716628), the French Society of Cardiology through Fondation “Coeur et Recherche,” “La Fédération Française de Cardiologie”, and Fondation pour la Recherche Medicale (FRM-2023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors confirm that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yu, M., Bouatia-Naji, N. Insights into the Inherited Basis of Valvular Heart Disease. Curr Cardiol Rep 26, 381–392 (2024). https://doi.org/10.1007/s11886-024-02041-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-024-02041-6