
Abstract
Purpose of Review
Immune cells are emerging as central cellular components of the heart which communicate with cardiac resident cells during homeostasis, cardiac injury, and remodeling. These findings are contributing to the development and continuous expansion of the new field of cardio-immunology. We review the most recent literature on this topic and discuss ongoing and future efforts to advance this field forward.
Recent Findings
Cell-fate mapping, strategy depleting, and reconstituting immune cells in pre-clinical models of cardiac disease, combined with the investigation of the human heart at the single cell level, are contributing immensely to our understanding of the complex intercellular communication between immune and non-immune cells in the heart. While the acute immune response is necessary to initiate inflammation and tissue repair post injury, it becomes detrimental when sustained over time and contributes to adverse cardiac remodeling and pathology.
Summary
Understanding the specific functions of immune cells in the context of the cardiac environment will provide new opportunities for immunomodulation to induce or tune down inflammation as needed in heart disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart contractility is compromised in the deadly syndrome of heart failure (HF). The full repertoire of cardiac cells and their gene expression profiles have been well characterized in the human and mouse heart at the single cell and single-nucleus level. While cardiomyocytes, accounting for roughly 30% of all cardiac cells, are the primary source of power for cardiac contractility, non-myocyte cells in the heart are also required for cardiac physiology [1,2,3•, 4]. Among the non-myocyte cells, different types of immune cells change in number in response to a variety of cardiac insults by infiltrating and/or expanding within the heart. Additionally, resident and infiltrating immune cells communicate with myocytes and other non-myocytes in the heart through soluble messengers and through direct cell contact and contribute to cardiac pathophysiology.
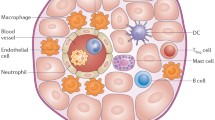
The immune system has evolved as a protective mechanism against pathogens. A coordinated response between its different cellular components is also required to distinguish between foreign and self, to clear cell debris, and promote tissue repair in response to sterile injury. Innate immune cells derive from a common myeloid progenitor (CMP) and are the body’s first and rapid response to injury and to infection. Mast cells, neutrophils, monocytes, and macrophages have been reported in the heart and play different roles in cardiac homeostasis and repair [3•]. The adaptive immune system, comprised of different subsets of T and B cells that derive from a common lymphoid progenitor (CLP), participate in a slower yet specialized response that requires antigen recognition, and results in their clonal expansion and B cell antibody production. Bridging the gap between these two systems are dendritic cells (DCs), with the unique ability to initiate specific T cell responses through antigen presentation via the major histocompatibility complex (MHC), expressed on DC, to the T cell receptor (TCR). Along with the presence of these immune cells, natural killer cells, derived from CLPs but part of the innate immune response due to their inability to recognize antigens, have also been reported in the heart (Fig. 1) [2, 4]. The roles of these distinct types of immune cells during cardiac homeostasis, acute and chronic inflammation in scar formation during cardiac repair, as well as in adverse cardiac remodeling, are all areas of active investigation reviewed here.

A simplified overview of innate and adaptive immune cells. The innate immune system consists of a wide variety of cell types which derive from a common myeloid progenitor in the bone marrow. Innate immune cells are the first responders for host defense and respond to pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively). Innate immune cells include granulocytes (neutrophils, basophils, mast cells), phagocytes (monocytes and macrophages), natural killer (NK) cells, and dendritic cells (DC) which bridge the gap with the adaptive immune system, composed by T cells and B cells, derived from the common lymphoid progenitor and developed in the thymus. They populate peripheral lymphoid organs—spleen and lymph nodes—where they expand in response to antigens presented by DCs to carry out functions with specificity and memory. CD8+ and CD4+ naïve T cells become effector CD8+ cytotoxic T cells, and CD4+ T helper or regulatory cells upon antigen engagement. CD4+ further differentiate into T helper (Th) and T regulatory (Treg) cells, characterized by specific signature transcription factor expression and cytokine production. Th type 1 (Th1) cells express Tbet and produce the pro-inflammatory cytokine IFNγ. Th type2 (Th2) cells express GATA3 and release IL-4 and IL-13. Th type 17 (Th17) expresses RORγT and releases IL-17. And Tregs express the signature transcription factor FoxP3 and produce TGF-β and IL-10 that suppress T cell effector function. CD8+ cytotoxic T cells follow a similar Tc1, Tc2, and Tc17 differentiation (not shown), while B cells are responsible for producing antibodies in response to antigens. This schematic shows immune cells which have been reported in the heart. Created with BioRender.com
At the steady state, the healthy heart contains DCs, neutrophils, mast cells, and T cells, along with distinct macrophage populations that confer unique structural and electrical properties to the heart [2, 3•, 4]. Among macrophages, a subset of CC motif chemokine receptor 2 (CCR2−) resident macrophages is seeded into the heart during embryogenesis and maintained through local proliferation. These are either partially or fully replaced by CCR2+ circulating monocytes [3•, 5]. Cardiac resident macrophages (mTMs) maintain cardiac homeostasis by modulating electrical conduction through direct crosstalk with cardiomyocytes [6]. They patrol vessels through direct communication with cardiac endothelial cells using the vascular homing receptor chemokine (C-X3-C motif) receptor 1 (CX3CR1) and the lymphocyte function-associated antigen 1 (LFA-1) integrin, and this way prevents undesired leukocyte infiltration. They also facilitate debris clearing maintaining this way a cardiac immune quiescent environment state [6, 7]. Cardiac mast cells are also present in low numbers in the hearts of healthy C57/BL/6 mice and in the healthy human myocardium, as indicated by histological staining with toluidine blue [8], yet their role at steady state has not been clearly defined to date. A subset of CD4+ T cells, regulatory T cells (Tregs), widely recognized as suppressors of the immune response to maintain immune cell tolerance, are also critical in the maintenance of immune homeostasis [9, 10].
Here, we review the most recent findings related to cardiac acute and chronic immune responses in the context of cardiac repair and remodeling (Fig. 2). We focus on how specific immune cell populations shift in the heart and lymphoid organs in response to distinct cardiac insults that lead to adverse remodeling in two distinct etiologies of HF. We highlight the findings reported in experimental animal models that have guided the mechanistic understanding of cardiac immune responses and discuss how they may translate to clinical observations in the human heart and may be considered for immunomodulation in HF.

Immune cells in cardiac injury repair and remodeling. Created with BioRender.com
Immune Cells in Injury Repair
Cardiac inflammation and fibrosis are two important hallmarks of HF. The acute immune response triggered by injury is necessary to initiate cardiac repair and is indispensable for survival [11]. It co-exists with the fibrotic response and is required for extracellular matrix (ECM) deposition by cardiac fibroblasts. This step is needed to replace damaged and dead cardiomyocytes, a process involved in nearly all forms of heart disease [12]. However, given the limited ability of the heart to regenerate, excessive fibrosis and persistent inflammation are detrimental for cardiac physiology long term.
Innate Immune Cells in Ischemic Cardiac Repair
Ischemic heart disease as a result of myocardial infarction (MI), results in permanent loss of cardiac tissue and exemplifies well the importance of immune responses to repair the injured heart [13]. The molecular and cellular mechanisms involved have been mainly characterized in pre-clinical experimental models that induce permanent myocardial ischemia (MI), as well as myocardial ischemia followed by reperfusion (I/R) to better mimic the patient population. In mouse models, this response is comprised of an initial acute inflammatory phase that takes place within hours and days post ischemia, a proliferative phase which takes days to a week, and a remodeling phase that usually takes weeks post ischemia [14].
Shortly after ischemic injury, a coordinated innate immune response is initiated in response to danger-associated molecular patterns (DAMPs) released by dying cardiomyocytes and cardiac macrophages. Resident macrophages and cardiomyocytes release pro-inflammatory cytokines and chemokines such as interleukin 6 (IL-6), tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), CXC motif chemokine ligand 8 (CXCL8), and CC motif chemokine ligand 2 (CCL2) [15, 16]. These initiate the recruitment of circulating neutrophils and CCR2+ monocytes to the heart [17], which are mobilized to the heart from the bone marrow and the spleen to clear debris and further activate endothelial cells during the first 3 days post injury (Fig. 2) [17, 18•]. Neutrophils produce IL-6 and further activate endothelial cell production of CCL2, and the surface expression of intercellular adhesion molecule 1 (ICAM-1), which in turn promotes lymphocyte antigen 6 complex, locus 1 (Ly6Chi) CCR2+ monocyte recruitment to the heart [11, 19, 20]. Cardiac infiltrated neutrophils additionally promote macrophage polarization towards reparative phenotypes through the neutrophil gelatinase–associated lipocalin [21]. The critical importance of neutrophils in the acute inflammatory response to ischemia and cardiac repair is supported by studies in which neutrophil depletion before ischemia through intraperitoneal (i.p.) injection of Ly6G antibody resulted in a significant reduction of left ventricular ejection fraction after echocardiography and increased collagen deposition compared to isotype control i.p. injection at 1 and 2 weeks post infarction [21]. Recently, a subset of immature neutrophils has been shown to be expanded in the circulation of patients within 24–72 h after myocardial infarction (AMI) and to correlate with T cell pro-inflammatory responses. Because myocardial tissue of these patients or cardiac imaging was not reported, it remains unknown if this neutrophil subset participates in cardiac repair, or if, alternatively, may be used as a biomarker of persistent chronic inflammation [22]. Lastly, mast cells have also been reported to accumulate in the heart, peaking at day 7 post-MI, and modulating cardiomyocyte contractility via PKA-regulated Ca2+ interactions [23], and release pro-inflammatory mediators such as TNFα, to initiate a cascade with resident macrophages, endothelial cells, and subsequent infiltrating neutrophils [24].
This highly inflammatory phase is a consequence of a rapid and coordinated acute innate immune response and is followed by a reparative phase, characterized by the cardiac presence of Ly6Clow macrophages. Fate mapping and single-cell RNA-sequencing (scRNA-seq) studies have shown distinct populations of infiltrating monocytes into the injured myocardium to differentiate into cardiac resident macrophages between days 2 and 4 after infarction [5]. The activated vascular endothelium continues its release of CCL2, attracting more Ly6Chi and Ly6Clow monocytes to the site of injury. During the reparative phase, accumulated Ly6Chi monocytes differentiate into macrophages with a homeostatic phenotype, a process partially dependent on the nuclear receptor subfamily 4 group A member 1 (NR4A1) hormone, essential in Ly6Clow monocyte development [20]. This was corroborated in studies using Nr4a−/− mice showing increased infiltration of pro-inflammatory monocytes into the myocardium and compromised cardiac function and fibrosis after one-week post-MI compared to WT mice [25]. More studies support that an exquisite balance in the transition from the innate acute inflammatory phase to the proliferative phase is needed not only to avoid cardiac rupture, but also to mitigate adverse effects on cardiac remodeling long term that will negatively impact cardiac physiology. As an example, pharmacological inhibition and genetic deletion of the DAMP S100A9 were detrimental for cardiac function 7 and 21 days post-MI, as these interventions impair the transition of pro-inflammatory macrophages to pro-reparative macrophages [26]. Further support of the necessity of this switch towards repair mediated by pro-reparative macrophages is the novel role for cardiac basophils, another innate immune cell (Fig. 1) which secretes the pro-fibrotic cytokines IL-4 and IL-13 that additionally contribute to control Ly6Clow monocyte-macrophage transformation. In AMI patients, low basophil counts are accompanied by worsened cardiac outcomes and increased fibrosis [27], supporting a potential role for this understudied immune cell type in cardiac repair (Fig. 2). Although most of the mechanistic characterization of the acute innate immune response to myocardial ischemia arises from studies using permanent ischemia, the requirement of this response is also true for cardiac repair in ischemia followed by reperfusion (I/R), with the main distinction being the greater magnitude, but shorter duration of immune cell infiltration in I/R compared to the non-reperfused MI hearts [28]. A recent study in pigs also supports a role of inflammation in cardiac repair and contractile function in I/R and highlights the need of studying inflammatory mechanisms in large animals prior to transitioning to cardiac repair therapies in patients [29].
Adaptive Immune Cells in Ischemic Cardiac Repair
Alongside the innate immune cells, T cells also contribute to the early cardiac repair in response to ischemic injury. Early studies identified effector and Treg cells present in the heart early post-MI. These were followed by studies showing that CD4+ T cell–deficient mice, using Cd4−/− and Mhc-II−/−, had higher mortality due to cardiac rupture than wild-type controls. Moreover, impaired collagen deposition is seen after Ab-mediated CD4+ T cell depletion and Cd4−/− mouse models [30, 31]. The fact that mice with T cell receptors restricted to an exogenous peptide fragment of chicken ovalbumin, and thus unable to respond to endogenous antigens, had similar mortality than WT mice suggested that a cardiac neoantigen-specific T cell response was required for cardiac repair [30, 32]. More recently, some of these neoantigens triggering a T cell reparative response have been identified using a screening of MHC-II restricted epitopes in Balb/c mice with a TCR specific to a cardiac myosin peptide. T cells specific for the cardiac myosin peptide - myosin heavy chain α (MYHCA) - selectively accumulated in the heart and mediastinal draining lymph nodes and acquired a pro-repair Treg phenotype through indirectly inducing collagen deposition in the heart [33•]. Interestingly, myocardial Tregs were also detected in autopsy samples from AMI patients who showed mediastinal lymph node alterations that correlated with infarct size and cardiac function. More recently, a peptide fragment of the beta-1 adrenergic receptor (ADRB1) was found to elicit CD4+ T cell responses after MI in patients [34]. Although it is possible that other antigens besides these ones are required for cardiac repair, these studies support a central role of antigen-specific T cells in cardiac repair by modulating monocyte and cardiac fibroblast activation into reparative/pro-fibrotic states that prevent cardiac rupture. All CD4+ T cell effector subsets, along with CD8+ T cells, are found in the heart shortly after ischemia, but evidence shows that Tregs seem to dominate this reparative response [17, 30, 31, 35]. This is further supported by studies using CCL17-deficient (Ccl17−/−) mice, a chemokine predominantly produced by CCR2+ innate cells, which show increased Tregs in the ischemic heart and improved ventricular remodeling and function post ischemia (Fig. 2) [36].
Another subset of adaptive immune cells that populates the heart post-MI are B cells. As seen with scRNA-seq, flow cytometry, and histological analyses, B cells express CXCR5, the receptor for CXCL13, and infiltrate the heart 1 day post-MI via the CXCL13-CXCR5 axis, and peak at day 7 post-MI [37], where they contribute to local TGF-β1 production, possibly communicating with cardiac fibroblasts. Antibody-mediated neutralization of CXCL13 in mice and global deficiency of CXCR5 (Cxcr−/−) mice impaired B cell recruitment to the heart and cardiac Tgfβ1 expression but had no impact on contractile function (Fig. 2) [37]. More comprehensive studies are needed to better understand B cell immunity in the heart. These gaps include the identification of B cell subsets involved in cardiac repair and whether, and to what extent, their presence in the heart, or systemically, contributes to the production of antibodies to cardiac antigens post ischemic injury.
Innate and Adaptive Immune Cells in Adverse Remodeling After Cardiac Repair Post Ischemia
The cardiac repair process culminates in scar formation necessary for survival. However, sustained immune and fibrotic responses not limited to the site of injury continue to take place. These contribute to adverse cardiac remodeling in areas remote to the scar to compensate for the healed but unfortunately dysfunctional, and with limited regenerative potential, myocardium [38]. Not surprisingly, both innate and adaptive immune cells co-exist in the heart and lymphoid organs at this stage in a complex environment once the scar has been formed post ischemia. Studies in mice 8 weeks post-MI have shown increased frequency of macrophages with pro-inflammatory phenotypes, accompanied by substantial increases in fibrosis, as well as left ventricular (LV) systolic dysfunction. Moreover, this was associated with splenic remodeling, characterized by increased size of the spleen and an expansion of macrophages and lymphocytes in the marginal zone that suggest enhanced antigen processing and T cell activation [39]. The importance of the spleen in adverse cardiac remodeling post-MI was demonstrated in studies in which splenectomy resulted in improved LV remodeling associated with diminished cardiac infiltration of immune cells 8 weeks post-MI. This discovery demonstrated a novel cardio-splenic axis in ischemic HF (Fig. 2) [39].
Moreover, at this chronic time point post-MI, there is a robust expansion of CD4+ T cells in the circulation, the spleen, and the mediastinal lymph nodes, in addition to CD4+ T cell infiltration in the heart, including Treg cells [17, 40•]. Classically considered as suppressors of pro-inflammatory T cell responses necessary for their homeostatic immune tolerance functions [41], during chronic inflammation, Tregs can be plastic and lose their suppressive fitness. This was investigated in cardiac remodeling post ischemia in studies of selective ablation of Tregs using the Foxp3-diphtheria toxin (Foxp3-DTR) mice. Tregs were depleted 4 weeks post-MI, when cardiac fibrosis and LV dysfunction were evident, and followed for a total of 8 weeks that demonstrated improved LV function and decreased fibrosis compared to control mice which had not been depleted of Tregs. This finding demonstrated that Tregs were potentially pathogenic in ischemic cardiomyopathy, and uncovered their unique ability to adopt a pathogenic phenotype in chronic ischemic HF, characterized by the release of IFNγ and TNFα, impairing angiogenesis in the heart (Fig. 2) [40•].
Taken together, a wealth of elegant studies demonstrate that coordinated immune and fibrotic responses are necessary for cardiac repair and remodeling. Shifts in immune cell and cardiac fibroblast states dominate a chronic response that is maladaptive for the heart and contributes to ischemic HF. New investigations to understand the cellular interplay in the heart in the context of ischemic injury are warranted to develop effective immunomodulatory therapies for cardiac repair and non-ischemic HF.
Immune Cells in Averse Cardiac Remodeling in Response to Non-ischemic Cardiac Insults
Non-ischemic HF is a consequence of a wide range of conditions that include, but are not limited to, vascular and cardiac abnormalities that exert stress on the myocardium and result in adverse myocardial remodeling. Classic hallmarks of adverse remodeling include increase in cardiomyocyte size, chamber dilation, and cardiac fibrosis to adapt to higher filling pressures in the heart. These, in patients, correlate with systemic and cardiac increases of pro-inflammatory cytokines and pro-inflammatory innate and adaptive immune cells. Our knowledge about cardiac immune responses and their contribution to adverse remodeling in non-ischemic HF is exponentially growing with the use of pre-clinical experimental models combined with the in-depth characterization of human plasma and myocardial tissue. Transverse aortic constriction (TAC) induces abrupt pressure overload (PO) in the LV and overtime mimics the pathological hallmarks of HF seen in patients [42], allowing for the characterization of the special and temporal cellular immune responses reviewed herein.
Innate Immune Cells in Non-ischemic HF
As opposed to MI, non-ischemic HF is not initiated by immediate cardiomyocyte death. Instead, building pressure in the LV and mechanical stress results in the release of pro-inflammatory cytokines and DAMPs by damaged cells. Sensitive to the mechanical stimuli, cardiac myocytes initially respond through increased intracellular Ca2+ and calcium-calmodulin pathways, regulating hypertrophic signaling and cell growth [43, 44]. This leads to the activation of the NOD-like receptor protein 3 (NLRP3) inflammasome in cardiomyocytes, as shown in studies demonstrating that cardiomyocyte sensing of hemodynamic stress provokes inflammatory signals through Ca2+/calmodulin-dependent kinase protein kinase II (CaMKIIδ) that result in the activation of NLRP3 inflammasome [45]. Camkiiδ−/− mice showed decreased cardiac gene expression of the pro-inflammatory cytokines Il-1β, Il-6, and Tnfα and the chemokines Ccl2 and Ccl3 as early as 3 days post TAC, compared to WT mice. This work positioned cardiac myocytes as essential players in initiating inflammatory responses to PO [42].
With the simultaneous release of IL-1β and TNFα, ICAM-1 is expressed in the vascular endothelium as early as 48 h post TAC and is sustained over time through 4 weeks post TAC. A consequence of this is the extravasation of innate immune cells into the heart shortly after TAC. Indeed, Icam-1−/− mice have decreased innate cell and T cell infiltration as compared to WT mice in response to 4 weeks TAC [46]. Neutrophils, monocytes, macrophages, and dendritic cells infiltrate the heart in response to hemodynamic stress as early as 1 week post TAC, as reported in studies using flow cytometric characterization of Ly6ChiCCR2+ and Ly6ClowCX3CR1 monocytes and cardiac gene expression of Ccl2, Ccl7, and Ccl12 chemokines that suggest a signal that attracts this early wave of innate immune cells to the heart [47]. The importance of CCR2+ monocytes in adverse cardiac remodeling was demonstrated in studies using a small-molecule CCR2 antagonist as well as depleting CCR2+ monocytes with a monoclonal antibody, both of which resulted in preserved cardiac function and decreased cardiac inflammation, complimented with a decrease in T cell expansion in the mLN [47,48,49]. A recent study using scRNA-seq of cardiac leukocytes isolated 4 weeks post injury also supports the presence of mast cells in the heart as potential contributors to adverse cardiac remodeling [3]. The large infiltration of innate immune cells shortly after TAC (1 and 2 weeks) occurring simultaneously with the progression of cardiomyocyte hypertrophy supports a potential role for innate cells in the compensatory hypertrophy that precedes the pro-fibrotic remodeling. However, CCR2+ and CCR2− macrophages, as well as DCs, are also present in the heart at later time points post TAC and are thought to also contribute to the fibrotic response later on (Fig. 2) [50].
Adaptive Immune Cells in Non-ischemic HF
As chronic pressure overload increases, collagen deposition becomes more evident along with additional recruitment of adaptive immune cells. The dominant role for T cells in adverse cardiac remodeling was demonstrated in T cell–deficient mice. Tcr−/− mice, deficient in alpha beta T cells through deletion of the T cell receptor, and Mhc-II−/− mice, deficient in CD4+ T cells specifically, did not develop cardiac fibrosis, cardiac hypertrophy, or systolic dysfunction in response to TAC, in contrast to Cd8−/− mice, which had a similar phenotype to WT control mice. These studies positioned CD4+ T cells as responsible for adverse cardiac remodeling in response to TAC [51, 52]. Specifically, the CD4+IFNγ+ T cell subset (Th1 cells) was expanded in the mediastinal lymph nodes, and increased Ifng transcripts were observed in the heart 4 weeks post TAC. The functional necessity of Th1 cells in cardiac fibrosis was demonstrated in adoptive transfer studies of CD4+IFNγ+ and CD4+IFNγ− effector T cells into Tcra−/− mice which showed that only CD4+IFNγ+ T cells were able to induce cardiac fibrosis in Tcra−/− in the onset of TAC [53]. In vitro mechanistic studies further demonstrated that T cells isolated from mediastinal lymph nodes of TAC mice adhered to cardiac fibroblasts and induced their transformation to myofibroblasts, a process which was inhibited by blocking T cell α4 integrin–cardiac fibroblast VCAM-1 interactions.
Interestingly, during this intimate contact, cardiac fibroblasts express MHC-II induced by IFNγ and present antigen to T cells, as a way to induce T cell activation within the heart [54••]. In fact, using the TCR activation reporter mice (Nur77GFP mice), demonstrated that antigen engagement occurs in the heart during injury [55]. These data were further supported by studies using cardiac fibroblast–specific MHC-II-deficient mice (Tcf21iCreMhcIIfl/fl), which had reduced fibrosis and preserved systolic function in response to TAC [54••]. In support of CD4+IFNγ+ T cells dominating the remodeling response to cardiac pressure overload, a study reported that deficiency in T-box protein (T-bet), the Th1 cell signature transcription factor, resulted in decreased cardiac fibrosis and hypertrophy in rats in response to pressure overload (Fig. 2) [56].
Histological data also demonstrated that cardiac T cells are found infiltrated in the hearts of patients with end-stage non-ischemic HF, and that the majority of them express the chemokine receptor CXCR3 [50, 52]. This, together with the fact that T cells from HF patients have a higher affinity to adhere to endothelial cells than those from healthy controls, suggests enhanced T cardiotropism in HF patients [51]. Studies in Cxcr3−/− mice and in CXCL9/CXCL10 reporter mice further demonstrate that CXCL9 and CXCL10 produced by cardiac fibroblasts, monocytes, and macrophages over the course of TAC attract Th1 cells to the heart in a CXCR3-dependent manner [50]. The T cell antigens involved in response to hemodynamic stress are different from those identified in response to ischemia, and involve cardiac neoantigens formed in response to reactive oxygen species (ROS). ROS induces the formation of isolevuglandins (IsoLGs), which adduct to cardiac proteins which are presented by DCs to T cells. Indeed, scavenging of IsoLGs as well as treatment with anti-oxidants in mice prevents T cell activation and clonal expansion and the development of systolic function in response to TAC [55].
Taken together, the mechanistic investigations in mouse models of cardiac PO, combined with the presence of T cell alterations in humans with HF, support coordinated and sequential innate and adaptive immune responses that contribute to adverse cardiac remodeling and contractile dysfunction in non-ischemic HF conditions.
Concluding Remarks
The cardio-immunology field is progressing at a fast pace since the discovery that cardiac inflammation is present in HF patients in addition to the systemic inflammation that had been observed for decades. With the access to new technologies, human cardiac tissue, and experimental animal models of HF, the field has significantly grown to the point where it is now well accepted that immune cells are central to cardiac homeostasis and disease progression. The discovery of several immune cell types in the heart that contribute differently to cardiac repair and remodeling is raising intriguing and exciting questions about the potential to specifically modulate these responses in a timely and context-specific manner to optimally achieve immunomodulation in cardiac disease. A wealth of information now supports that the trigger of the cardiac insult determines the type of cardiac immune response, which, in turn, impacts cardiac pathophysiology. A lot of work needs to be done to continue identifying cardiac antigens that differentially modulate repair vs remodeling in response to different cardiac insults, as well as those that “tolerize” the heart against pathogenic immune invasion during homeostasis. These will help us understand why, for instance, immune check point inhibitors sometimes induce fatal inflammation in cancer patients, and help identify those who may be more at risk [57]. Additionally, identifying antigens will help develop specific T cell targeted therapies, as recently described with chimeric antigen receptor T cells (CAR-T cells) able to target fibrosis in experimental animal models [58]. Similarly, we are starting to understand sex differences in cardiac remodeling. Mouse studies suggest improved survival and limited remodeling after MI in female mice [59], and worsened outcomes in cardiac remodeling under the influence of acute cigarette smoke exposure post-MI in males compared to females [60]. Altogether, this calls for a sense of urgency to better understand sex differences in cardiac immune responses. Several reports reviewed here support a role for innate and adaptive immune cells in systolic and diastolic dysfunction in experimental models of HF with reduced ejection fraction. However, much work remains to be done to understand whether specific immune actions modulate contractility vs relaxation, something which may shed new light into how to treat HF with reduced ejection fraction (HFrEF) vs HF with preserved ejection fraction (HFpEF), a condition with limited effective treatments to date. Lastly, efforts must be made to understand the immune cell compartments during early stages of life, when the heart is able to regenerate. The finding that adult T cells transferred to neonate mice blunt the heart’s regenerative response supports that the cardio-immunology axis can be further explored in this arena. The more we continue learning about the immune-cardiac axis in different etiologies of HF, the closer researchers will be to the ultimate goal of finding new targets for immunomodulation of cardiac repair and remodeling.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Johnson EK, Matkovich SJ, Nerbonne, JM. Regional differences in mRNA and lncRNA expression profiles in non-failing human atria and ventricles. Sci Rep. 2018;8.
Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL et al. Cells of the adult human heart. Nature. 2020;588:466-+.
• Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. 2019;140:2089–107. This work delineated how immune cells in the myocardium change in response to cardiac pressure overload in mice through single cell sequencing and high-resolution analyses.
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R et al. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–9.
Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29-+.
Moskalik A, Niderla-Bielinska J, Ratajska A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail Rev. 2022;27:1413–30.
Pinto AR, Godwin JW, Rosenthal NA. Macrophages in cardiac homeostasis, injury responses and progenitor cell mobilisation. Stem Cell Research. 2014;13:705–14.
Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 2002;118:41–9.
Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z et al. Foxp3(+)CD25(+)CD4(+) natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27.
Xia N, Lu YZ, Gu MY, Li NN, Liu ML, Jiao J et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142:1956–73.
Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction from inflammation to fibrosis. Circ Res. 2016;119:91–112.
Liu T, Song D, Dong JZ, Zhu PH, Liu J, Liu W et al. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front Physiol. 2017;8.
Sadek H, Olson EN. Toward the goal of human heart regeneration. Cell Stem Cell. 2020;26:7–16.
Nunes-Silva V, Frantz S, Ramos GC. Lymphocytes at the heart of wound healing. Adv Exp Med Biol. 2017;1003:225–50.
Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res. 2004;53:585–95.
Frangogiannis NG, Dewald O, Xia Y, Ren GF, Haudek S, Leucker T et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–92.
Yan XX, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35.
• Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. This work identified a splenic reservoir of monocytes that are mobilized to the heart post ischemia.
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24:1234-+.
Hilgendorf I, Gerhardt LMS, Tan TC, Winter C, Holderried TAW, Chousterman BG et al. Ly-6C(high) monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–22.
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–97.
Fraccarollo D, Neuser J, Moller J, Riehle C, Galuppo P, Bauersachs J. Expansion of CD10(neg) neutrophils and CD14(+)HLA-DRneg/low monocytes driving proinflammatory responses in patients with acute myocardial infarction. Elife. 2021;10.
Ngkelo A, Richart A, Kirk JA, Bonnin P, Vilar J, Lemitre M et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med. 2016;213:1353–74.
Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710.
Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C(-) monocytes. Nat Immunol. 2011;12:778-U148.
Marinkovic G, Koenis DS, de Camp L, Jablonowski R, Graber N, de Waard V et al. S100A9 links inflammation and repair in myocardial infarction. Circ Res. 2020;127:664–76.
Sicklinger F, Meyer IS, Li X, Radtke D, Dicks S, Kornadt MP et al. Basophils balance healing after myocardial infarction via IL-4/IL-13. J Clin Investig. 2021;131.
Vandervelde S, van Amerongen MJ, Tio RA, Petersen AH, van Luyn MJ, Harmsen MC. Increased inflammatory response and neovascularization in reperfused vs. non-reperfused murine myocardial infarction. Cardiovasc Pathol. 2006;15:83–90.
Bonner F, Gastl M, Nienhaus F, Rothe M, Jahn A, Pfeiler S et al. Regional analysis of inflammation and contractile function in reperfused acute myocardial infarction by in vivo (19)F cardiovascular magnetic resonance in pigs. Basic Res Cardiol. 2022;117:21.
Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G et al. Activation of CD4(+) T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652-U146.
Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G et al. Lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail. 2017;10.
Weirather J, Hofmann UDW, Beyersdorf N, Ramos GC, Vogel B, Frey A et al. Foxp(3+) CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67.
• Rieckmann M, Delgobo M, Gaal C, Buchner L, Steinau P, Reshef D et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Investig. 2019;129:4922–36. This work identified cardiac myosin as a dominant cardiac antigen within the injured myocardium after myocardial infarction.
Hapke N, Heinrichs M, Ashour D, Vogel E, Hofmann U, Frantz S et al. Identification of a novel cardiac epitope triggering T-cell responses in patients with myocardial infarction. J Mol Cell Cardiol. 2022;173:25–9.
Kumar V, Prabhu SD, Bansal SS. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front Cardiovasc Med. 2022;9.
Feng GS, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation. 2022;145:765–82.
Heinrichs M, Ashour D, Siegel J, Buchner L, Wedekind G, Heinze KG et al. The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc Res. 2021;117:2664–76.
Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18:733–44.
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure. Circ Res. 2014;114:266–82.
• Bansal SS, Ismahil MA, Goel M, Zhou GH, Rokosh G, Hamid T et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. 2019;139:206–21. This work provided evidence of dysfunctional cardiac Tregs as pro-inflammatory and essential drivers of pathology in chronic ischemic heart failure.
Sakaguchi S, Vignali DAA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. 2013;13:461–7.
Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138:2530–44.
Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49.
Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Investig. 2000;105:1395–406.
Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol. 2019;16:361–78.
Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang BN, Molina AA et al. Intercellular adhesion molecule 1 regulates left ventricular leukocyte infiltration, cardiac remodeling, and function in pressure overload-induced heart failure. J Am Heart Assoc. 2016;5.
Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M et al. CCR2(+) Monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. Jacc-Basic to Translational Science. 2018;3:230–44.
Liao XD, Shen YY, Zhang RL, Sugi K, Vasudevan NT, Alaiti MA et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc Natl Acad Sci USA. 2018;115:E4661–9.
Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD. Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. Plos One. 2017;12.
Ngwenyama N, Salvador AM, Velazquez F, Nevers T, Levy A, Aronovitz M et al. CXCR3 regulates CD4(+) T cell cardiotropism in pressure overload-induced cardiac dysfunction. Jci Insight. 2019;4.
Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C et al. CD4(+) T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111-+.
Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M et al. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circulation-Heart Failure. 2015;8:776-U160.
Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017;214:3311–29.
•• Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R et al. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res. 2022;1:761–74. This work identified that cardiac fibroblasts function as antigen-presenting cells that modulate T cell immune responses.
Ngwenyama N, Kirabo A, Aronovitz M, Velazquez F, Carrillo-Salinas F, Salvador AM et al. Isolevuglandin-modified cardiac proteins drive CD4+ T-cell activation in the heart and promote cardiac dysfunction. Circulation. 2021;143:1242–55.
Ma ZG, Dai J, Yuan YP, Bian ZY, Xu SC, Jin YG et al. T-bet deficiency attenuates cardiac remodelling in rats. Basic Res Cardiol. 2018;113.
Baik AH, Oluwole OO, Johnson DB, Shah NI, Salem JE, Tsai KK et al. Mechanisms of cardiovascular toxicities associated with immunotherapies. Circ Res. 2021;128:1780–801.
Rurik JG, Tombacz I, Yadegari A, Fernandez POM, Shewale SV, Li L et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91-+.
Pullen AB, Kain V, Serhan CN, Halade GV. Molecular and cellular differences in cardiac repair of male and female mice. J Am Heart Assoc. 2020;9.
Kaplan A, Abidi E, Diab R, Ghali R, Al-Awassi H, Booz GW et al. Sex differences in cardiac remodeling post myocardial infarction with acute cigarette smoking. Biol Sex Differ. 2022;13.
Funding
This work was supported by NIH-R01 HL 144477 (to PA) and HL144477-S3 (to MZ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zambrano, M.A., Alcaide, P. Immune Cells in Cardiac Injury Repair and Remodeling. Curr Cardiol Rep 25, 315–323 (2023). https://doi.org/10.1007/s11886-023-01854-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-023-01854-1