
Abstract
Purpose of Review
Truncating TTN variants (TTNtv) are the most common genetic cause of dilated cardiomyopathy (DCM), but the underlying mechanisms are incompletely understood and effective therapeutic strategies are lacking. Here we review recent data that shed new light on the functional consequences of TTNtv and how these effects may vary with mutation location.
Recent Findings
Whether TTNtv act by haploinsufficiency or dominant negative effects has been hotly debated. New evidence now implicates both mechanisms in TTNtv-related DCM, showing reduced titin content and persistent truncated titin that may be incorporated into protein aggregates. The extent to which aggregate formation and protein quality control defects differ with TTNtv location and contribute to contractile dysfunction is unresolved.
Summary
TTNtv-associated DCM has a complex etiology that involves varying combinations of wild-type titin deficiency and dominant negative effects of truncated mutant titin. Therapeutic strategies to improve protein handling may be beneficial in some cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Titin is the largest human protein and the third most abundant myofilament in the sarcomere in heart and skeletal muscle. It is essential for normal sarcomerogenesis and has key roles in maintaining the structural stability of muscle cells and in active and passive sarcomere function. In the heart, titin also plays a vital role in responding to mechanical stress through its ability to recruit protein binding partners involved in mechano-signaling, calcium signaling, cardiac metabolism and protein quality control (PQC) [1, 2•]. Variants that truncate, or shorten, the titin protein (TTNtv) are the most common genetic cause of dilated cardiomyopathy (DCM) [3, 4]. However, there is significant variability in the age of onset and severity of DCM amongst TTNtv carriers, and TTNtv are also found in up to 3% of the general population [3, 4].
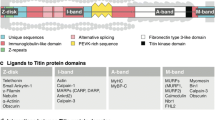
Much of this phenotypic variability has been accounted for by consideration of the position of each TTNtv. Early reports showed that DCM-associated TTNtv were mainly located in the A-band region of the titin protein (Fig. 1). While this is true, DCM-associated TTNtv do occur in other titin domains and A-band TTNtv are also present in the general population. There are several titin isoforms in the mature heart that differ mainly in the extent of alternative splicing in the I-band (Fig. 1). A major advance in evaluating TTNtv was the derivation of the proportion spliced in (PSI) scores for each titin exon that represent the frequency in which that exon is included across the range of titin isoforms. TTNtv located in highly used exons (defined by PSI > 0.9) were considered more likely to be pathogenic than those in exons with low scores [4]. Assessing exon PSI scores has become the cornerstone for clinical interpretation of TTNtv. However, questions remain about the impact of TTNtv location on disease severity, especially for variants outside the A-band region, and also why some TTNtv carriers remain asymptomatic throughout life. Studies in genetically modified zebrafish introduced a further hypothesis that the clinical manifestation of TTNtv was determined by variant location with respect to the promoter of a short titin isoform called Cronos in the distal I-band region (Fig. 1). It was proposed that variants occurring proximal to the Cronos promoter would have a relatively less severe phenotype due to upregulation of the Cronos isoform [5]. These rescue effects would not be possible for variants distal to the promoter that truncate both the full-length and Cronos titin isoforms. Although appealing, convincing evidence for this hypothesis in human heart studies has been lacking. It is generally agreed that phenotypic variability of TTNtv is incompletely explained by variant factors alone, and that unique combinations of background genetic and environmental factors are also involved in individual patients (Fig. 1).

Summary of established and newly proposed determinants of TTNtv pathogenicity. The top line shows a schematic of the full-length titin protein and its major domains: Z-line, I-band, A-band and M-line. There are three major titin isoforms expressed in adulthood: N2A (predominantly expressed in skeletal muscle), N2BA and N2B (main cardiac titin isoforms). These isoforms differ in composition mainly due to alternative splicing of I-band exons. The shorter Cronos isoform is transcribed from a promoter in the distal I-band. Currently, the major factor that has been used to identify deleterious TTNtv is variant location, i.e. in the A-band, distal to the Cronos promoter, or in exons with high proportion spliced-in (PSI) scores. New data suggest that titin haploinsufficiency is a consequence of all TTNtv, and additionally redefine variant pathogenicity by considering variant location with respect to the major I-band splicing region which may influence the abundance of truncated titin protein and aggregate formation. (Figure adapted from Fatkin and Huttner [6], with permission from Wolters Kluwer Health, Inc.)
The effects of TTNtv on the encoded titin protein have been unclear. Most adult patients are heterozygous for TTNtv, that is, they carry one normal ("wild-type") copy and one mutant copy of the TTN gene. There are two possible outcomes for the mutant allele. First, the abnormal messenger RNA (mRNA) transcript may undergo nonsense-mediated decay (NMD), generally resulting in a 50% reduction of the total amount of wild-type titin protein (haploinsufficiency). Alternatively, the mutant allele may be translated, leading to the expression of a truncated protein that persists and interferes with normal sarcomere function (dominant negative or “poison peptide” effects). Previous attempts to elucidate whether one or both of these mechanisms are at play in TTNtv-related cardiomyopathy have been inconclusive, with no clear data to show either reduced titin protein levels or the presence of truncated titin protein. Within this context, three recently published papers provide intriguing new perspectives on the pathogenesis of TTNtv-associated DCM and offer exciting new opportunities for therapeutic intervention [7••, 8••, 9•]. The highlights of these publications and directions for future research are outlined below.
Titin Haploinsufficiency and Reduced Sarcomere Content
Haploinsufficiency has been considered a key mechanism underpinning TTNtv pathogenicity, partly owing to a lack of evidence supporting the existence of truncated titin peptides [4, 10, 11]. However, several studies have found no evidence of NMD of TTN variant alleles [4, 7••, 8••, 12•, 13]. In fact, it has been shown that TTNtv, particularly N-terminal TTNtv, undergo inefficient termination of translation of the premature stop codon rather than NMD [12•].
Titin haploinsufficiency has been linked to abnormalities in sarcomerogenesis, resulting in fewer sarcomeres being formed during heart development. Studies undertaken in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM), engineered cardiac microtissues and zebrafish have demonstrated that TTNtv cause defects in sarcomere assembly and myofibrillogenesis, leading to reduced force generation and sarcomere content, termed "sarcomere insufficiency" [9•, 10, 14–16]. Despite this, attempts to demonstrate reduced titin mRNA and/or protein levels in human myocardial tissue and other TTNtv animal models have provided inconsistent results [9•, 10, 14, 15, 17]. This variability may be explained, at least in part, by the technical challenges of evaluating the giant titin protein using standard techniques.
In an elegant series of experiments, Fomin et al. [7••] used two-phase gels to quantify titin expression relative to other proteins in human heart tissues. When compared to idiopathic DCM (n = 91) or donor (n = 14) hearts, TTNtv hearts showed a substantial reduction in total titin protein [7••]. These results suggested that sarcomere insufficiency might be a distinct consequence of TTNtv rather than a non-specific effect of DCM. Interestingly, there was a parallel reduction in β-myosin heavy chain levels in the TTNtv hearts which could further contribute to sarcomere insufficiency. Evidence for reduced sarcomere content in TTNtv hearts was also gained by quantification of sarcomere numbers using immunostaining. In a co-published article, McAfee et al. [8••] reported heart tissue analyses in patients with high PSI TTNtv (n = 22), non-ischemic DCM (n = 158) and unused donor hearts (n = 14). Using RNA sequencing, they compared the ratio of wild-type to mutant TTNtv transcripts, reasoning that if NMD was occurring there would be a significantly higher amount of wild-type TTN transcripts compared to mutant transcripts (i.e. "allelic imbalance"). However, there was only modest evidence of allelic imbalance suggesting that TTNtv alleles did not undergo significant NMD. These authors went on to evaluate titin protein levels, finding a 30% reduction in the levels of full-length titin protein in TTNtv hearts when compared to non-TTNtv hearts. Levels of truncated titin were also reduced but no degradation products were detected. Collectively, these two new datasets provide compelling evidence for haploinsufficiency in TTNtv-associated DCM (Fig. 1).
Cronos Hypothesis Debunked?
The importance of upregulation of the Cronos titin isoform as a determinant of TTNtv manifestation has been unclear; however, recent data have provided clarity on this point. In the mature heart, expression levels of Cronos are low, representing only 2–3% of total cardiac protein and 12% of total titin protein [7••, 18•]. In their human tissue analyses, Fomin et al. [7••] found no differences in Cronos transcript or protein levels between TTNtv hearts, idiopathic DCM hearts and donor hearts. Moreover, Cronos expression was indistinguishable for TTNtv located proximal and distal to the Cronos transcriptional start site. These findings clearly show that Cronos does not compensate for loss of full-length titin isoforms or play a major role in adult-onset TTNtv-associated DCM.
Although Cronos may not be able to rescue adult-onset disease, it is known to be highly expressed during embryonic development where it is involved in the proper formation of sarcomeres, primarily in skeletal muscle but also in cardiomyocytes [5, 18•]. Previous work in zebrafish and hiPSC-CM suggests that Cronos can partially rescue sarcomere formation and function in settings of TTNtv homozygosity, where no functional full-length titin is produced [5, 18•]. Therefore, it is possible that Cronos might support compensated heart function early in life in heterozygous carriers of pre-Cronos TTNtv, mitigating against the subsequent development of DCM.
Position-dependent Expression of Truncated Titin?
Whether or not truncated titin proteins are expressed has been a topic of great debate for many years. Although truncated titin peptides have consistently been observed in in vitro models of TTNtv-associated DCM [9•, 14, 15], protein studies in human myocardial tissue, and rodent and zebrafish models have generally failed to detect truncated titin peptides [4, 10, 11, 13].
McAfee et al. [8••] and Fomin et al. [7••] have now provided robust new data to show that truncated titin proteins are expressed in the adult human heart. Using proteomics assays, McAfee et al. [8••] identified small titin peptide fragments from wild-type and TTNtv alleles, indicating that the mutant alleles were translated into protein. Subsequent gel electrophoresis and immunoblotting demonstrated a range of truncated titin proteins with intact N-terminal sequences but absent C-terminal sequences (Fig. 2A). It should be noted that all of the TTNtv in this study were in the distal I-band, A-band, or M-line regions.

Evidence of truncated titin protein expression observed in human myocardial tissue. (A) McAfee et al. [8••] observed a staircase-like pattern of bands corresponding to the expected size of truncated (tr-) N2BA (yellow arrowheads) and/or N2B (red arrowheads) titin peptides in the hearts of DCM patients with M- and A-band TTNtv. (From McAfee et al. [8••]. Reprinted with permission from AAAS.) (B) Fomin et al. [7••] observed a similar pattern of truncated titin (purple asterisks) on Western blots. However, elevated amounts of tr-titin (particularly tr-N2BA) were clearly observed in pre-splice, but not in post-splice TTNtv hearts. Faint bands corresponding to tr-N2B were observed in post-splice TTNtv hearts. Tr-Cronos was also observed at the expected size in all post-splice TTNtv samples investigated. Variability in the amount of tr-titin expressed in TTNtv hearts was observed in both studies. (From Fomin et al. [7••]. Reprinted with permission from AAAS.)
Fomin et al. [7••] also found clear evidence of truncated titin protein, particularly when they looked at cardiac tissue from patients with proximal Z-disc and I-band TTNtv that had relatively shorter and readily identifiable truncated titin proteins (Fig. 2B). There was a greater abundance of truncated proteins for TTNtv proximal to the heavily spliced region of the I-band than for TTNtv in the distal I-band, A-band and M-line, leading the authors to propose that looking at variant location proximal or distal to the I-band major splicing region, rather than the Cronos promoter site, might be a more informative way to assess variant effects (Fig. 1) [7••]. In contrast to these findings in adult heart tissues, Romano et al. [9•] observed truncated titin peptides in hiPSC-CM carrying an A-band TTNtv but not in those carrying an I-band TTNtv. These apparently discrepant findings could be related to experimental differences or individual variability in titin protein turnover and warrant further investigation.
Titin Protein Aggregates and Protein Quality Control Defects
How does truncated titin contribute to DCM pathogenesis? It has been proposed that truncated titin proteins act as "poison peptides" by integrating into the sarcomere but failing to form mature myofibrils, leading to impaired myofibril force generation [9•, 14, 15]. To address this, McAfee et al. [8••] used myocardial protein isolation to separate sarcomeric proteins from other soluble and membrane-bound proteins in the cell. After identifying truncated titin in the sarcomeric fraction, they concluded that truncated titin is indeed integrated into the sarcomere, but also noted that truncated titin was more likely to dissociate into the soluble fraction and may form aggregates [8••]. Fomin et al. [7••] also addressed this issue, instead using immunostaining of myocardial tissue sections stained with titin Z-disc or titin M-line antibodies, indexed to α-actinin staining (a sarcomeric Z-disc protein). As the M-line antibody only recognises full-length, and not truncated titin, they reasoned that if truncated titin is integrated into sarcomeres, the ratio of M-line to Z-disc titin should be lower in TTNtv hearts compared with idiopathic DCM hearts. However, as the ratio was indistinguishable between the two groups they concluded that truncated titin is not integrated into the sarcomere in meaningful amounts [7••].
Using immuno-electron microscopy, Fomin et al. [7••] further identified electron-dense areas consistent with intracellular aggregate formation. These aggregates stained positive for antibodies that recognized the titin Z-disc region but not for M-line antibodies, indicating that they contained only truncated, and not full-length titin [7••]. This is an intriguing finding since titin aggregates have not been previously recognized as characteristic of TTNtv-associated DCM. Whether more aggregates form in hearts with pre-splicing TTNtv, in line with the greater amounts of truncated titin observed by the authors, is unclear and warrants further investigation.
Fomin et al. [7••] went on to suggest that the accumulation of these titin aggregates could impair protein homeostasis by sequestering PQC machinery. Interestingly, they found that wild-type titin was highly ubiquitinated in TTNtv hearts compared to idiopathic DCM and healthy hearts. While hyper-ubiquitination of cardiac proteins has been previously observed in DCM hearts [19], these results suggest that TTNtv hearts are relatively more affected. In contrast, there was minimal evidence of ubiquitination of truncated titin peptides, suggesting that they fail to be targeted for degradation via the UPS. This is the first evidence to suggest that ubiquitin-dependent degradation is impaired in TTNtv carriers.
This was further associated with derangements in the levels of key markers of the UPS (MURF1, SQSTM1/p62), unfolded protein response (CRYAB) and autophagy (LC3B) pathways. McAfee et al. [8••] also evaluated the expression of numerous autophagy markers, including LC3B and p62, but found no significant differences between TTNtv-positive and TTNtv-negative hearts. Defects in autophagy have been observed in rodent models of TTNtv [20•, 21], but the latest findings in human tissues suggest that, overall, the autophagy pathway may be relatively less impaired than the UPS.
Altogether, these studies suggest that PQC defects could contribute to TTNtv-related DCM, but also raise a number of additional questions. Whether PQC defects are directly caused by the sequestration of titin aggregates or are a consequence of DCM is unclear. If PQC defects precede DCM, this could make them an important therapeutic target to prevent or delay disease onset. Additionally, post-translational modifications of titin are known to be a key determinant of cardiomyocyte passive stiffness and ventricular diastolic function [22]. How titin truncation and downstream PQC defects might influence these processes is unknown. Furthermore, although incompletely understood, current data suggests that some TTNtv produce more truncated titin than others, raising the intriguing possibility that TTNtv carriers may be affected by distinct pathologies that are, in part, governed by variant location.
TTNtv and Disease Modifiers
Additional genetic and environmental factors have long been thought to contribute to the phenotypic variability observed in TTNtv carriers. Indeed, there remains some debate as to whether such factors are required for DCM to manifest in TTNtv carriers. However, a growing body of work suggests that while TTNtv can be sufficient to cause DCM on their own, factors such as hemodynamic stress [10], alcohol excess [23], chemotherapy [24] and pregnancy [25] act as disease modifiers. The mechanisms underlying these interactions remain incompletely understood, but these findings suggest that gene-environment interactions play an important role in disease pathogenesis in TTNtv carriers.
Targeted Treatments for TTNtv-associated DCM
Current treatment of TTNtv-associated DCM consists of standard heart failure medications such as ACE-inhibitors and β-blockers which provide symptomatic relief but do not address the underlying disease etiology. Fomin et al. [7••] demonstrated that treatment with proteasomal inhibitors could partially rescue PQC defects and contractile function in hiPSC-CMs. For human patients, however, therapeutic strategies targeting the PQC may not be feasible as they would need to be highly specific in order to reduce undesired off-target effects.
Genome-editing techniques such as exon skipping have previously been proposed as a potential treatment for TTNtv [26, 27]. Exon skipping acts on the mutant RNA to splice out a mutation-containing exon, leading to a truncated but potentially normally functioning protein. This strategy has shown promise for other heritable cardiac and skeletal muscle disorders conditions such as Duchenne muscular dystrophy that are caused by an absence of functional protein [28]. In the context of TTNtv, however, exon skipping may have limited efficacy given the potential role of truncated proteins in disease pathogenesis. Fomin et al. [7••] also investigated the utility of genetic correction of TTNtv showing complete rescue of contractile dysfunction in hiPSC-CM in a highly targeted manner. Similarly, Romano et al. [9•] used gene-editing techniques in an A-band hiPSC-CM TTNtv model to restore the normal reading frame and full-length titin production. These in vitro findings must be validated in vivo, and progress in this area will likely bring to the fore practical and ethical concerns regarding genetic screening, editing and possible off-target effects.
Conclusions and Future Directions
Recent data that have provided novel insights into molecular mechanisms underlying TTNtv pathogenicity, showing that while haploinsufficiency might be the key player in disease onset, dominant negative effects that lead to the accumulation of truncated titin aggregates and subsequent PQC defects may be present in a subset of TTNtv carriers. These factors are not mutually exclusive and, when present together, could accelerate disease progression and/or increase disease severity. Exactly how and when titin aggregates form and how they contribute to DCM pathogenesis remains unclear. Overall, these findings form the basis for a new conceptual framework in which TTNtv-associated DCM is not a single disease. Instead, there could be distinctive pathogenetic mechanisms based on variant location with respect to titin's major splicing region (Fig. 1). These latest data add new dimensions to our understanding of TTNtv-associated DCM and suggest that personalised treatment approaches may be possible, and perhaps even necessary.
One important caveat of this recent work is that the heart tissues evaluated were obtained from patients with end-stage DCM and hence it is impossible to know whether the defects identified had a key role in causing DCM or arose as a consequence of the failing heart. Ideally, serial evaluation of heart tissue before and after DCM onset is needed. Such studies are not possible in human hearts and are challenging in rodent TTNtv models that generally do not develop DCM without additional stress [13, 17]. In contrast, zebrafish TTNtv models spontaneously show progressive DCM with increasing age [10], making them uniquely suited to mechanistic analyses.
Regardless of whether TTNtv fundamentally act by haploinsufficiency or dominant negative effects, the downstream pathways that culminate in myocardial contractile dysfunction remain incompletely understood. A number of factors could be involved, including reduced force generation or force transmission in cardiomyocytes [9•, 14, 15], abnormal responses to mechanical stress [10, 14], defective myocardial energetics [13, 20•, 29], or increased susceptibility to endogenous and exogenous factors that depress myocardial function. Elucidating key pathogenetic pathways and the full spectrum of genetic and environmental modifiers of TTNtv should allow preventative biologically-targeted therapies and nuanced approaches to clinical management.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Linke WA. Titin gene and protein functions in passive and active muscle. Annu Rev Physiol. 2018. https://doi.org/10.1146/annurev-physiol-021317-121234.
• Loescher CM, Hobbach AJ, Linke WA. Titin (TTN): from molecule to modifications, mechanics, and medical significance. Cardiovasc Res. 2021;1:16. A comprehensive review of titin’s functions and properties, with a focus on the latest proposed mechanisms of TTNtv pathogenicity.
Herman DS, Lam L, Taylor MRG, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28.
Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015. https://doi.org/10.1126/scitranslmed.3010134.
Zou J, Tran D, Baalbaki M, et al. An internal promoter underlies the difference in disease severity between N-and C-terminal truncation mutations of titin in zebrafish. Elife. 2015. https://doi.org/10.7554/eLife.09406.
Fatkin D, Huttner IG. Titin-truncating mutations in dilated cardiomyopathy: The long and short of it. Curr Opin Cardiol. 2017;32:232–8.
•• Fomin A, Gärtner A, Cyganek L, et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med. 2021. https://doi.org/10.1126/SCITRANSLMED.ABD3079. Findings from this study provided clear evidence that wild-type titin is reduced, and that truncated titin is expressed in TTNtv hearts and forms cytosolic aggregates. They uniquely proposed that titin aggregates lead to PQC defects.
•• McAfee Q, Chen CY, Yang Y, et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13:7287. Co-published with Fomin et al. [7] this study also detected truncated titin protein expression and reduced titin content in human myocardial tissue taken from TTNtv carriers with DCM.
• Romano R, Ghahremani S, Zimmerman T, Legere N, Thakar K, Ladha FA, et al. Reading frame repair of ttn truncation variants restores titin quantity and functions. Circulation. 2022;145:194–205. Findings from this study further support the existence of position-dependent effects of TTNtv, showing that A-band TTNtv lead to both haploinsufficiency and dominant negative effects, while I-band TTNtv lead only to haploinsufficiency.
Huttner IG, Wang LW, Santiago CF, et al. A-Band Titin truncation in zebrafish causes dilated cardiomyopathy and hemodynamic stress intolerance. Circ Genom Precis Med. 2018. https://doi.org/10.1161/CIRCGEN.118.002135.
Shih YH, Dvornikov A, Zhu P, Ma X, Kim M, Ding Y, et al. Exon-and contraction-dependent functions of titin in sarcomere assembly development. 2016. https://doi.org/10.1242/dev.139246
• van Heesch S, Witte F, Schneider-Lunitz V, et al. The translational landscape of the human heart cell. 2019;178:242–260.e29. This study provided some of the first evidence that truncated titin proteins are translated, and that some TTNtv may be more likely to produce truncated peptides than others.
Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017. https://doi.org/10.1038/ng.3719.
Hinson JT, Chopra A, Nafissi N, et al. (2015) Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 1979;349:982–6.
Chopra A, Kutys ML, Zhang K, et al. Force Generation via β-Cardiac Myosin, Titin, and α-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev Cell. 2018;44:87-96.e5.
Xu X, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, et al. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet. 2002. https://doi.org/10.1038/ng816.
Gramlich M, Michely B, Krohne C, et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol. 2009. https://doi.org/10.1016/j.yjmcc.2009.04.014.
• Zaunbrecher RJ, Abel AN, Beussman K, et al. Cronos Titin is expressed in human cardiomyocytes and necessary for normal sarcomere function. Circulation 2019;140:1647–1660. This study provided the first evidence that Cronos is expressed in adult human cardiomyocytes.
Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003. https://doi.org/10.1002/pmic.200390029.
• Zhou J, Ng B, Ko NSJ, et al. Titin truncations lead to impaired cardiomyocyte autophagy and mitochondrial function in vivo. Hum Mol Genet. 2019;28:197–1981. This study provided the first evidence that TTNtv lead to defects in autophagy.
Radke MH, Polack C, Methawasin M, Fink C, Granzier HL, Gotthardt M. Deleting full length titin versus the titin m-band region leads to differential mechanosignaling and cardiac phenotypes. Circulation. 2019;139:1813–27.
Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein titin’s role in cardiomyopathy: genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front Physiol. 2019. https://doi.org/10.3389/fphys.2019.01436.
Ware JS, Amor-Salamanca A, Tayal U, et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71:2293–302.
Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation. 2019;140:31–41.
Goli R, Li J, Brandimarto J, et al. Genetic and phenotypic landscape of peripartum cardiomyopathy. Circulation. 2021. https://doi.org/10.1161/CIRCULATIONAHA.120.052395.
Gramlich M, Pane LS, Zhou Q, et al. Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol Med. 2015;7:562–76.
Hahn JK, Neupane B, Pradhan K, Zhou Q, Testa L, Pelzl L, et al. The assembly and evaluation of antisense oligonucleotides applied in exon skipping for titin-based mutations in dilated cardiomyopathy. J Mol Cell Cardiol. 2019;131:12–9.
Takeda S, Clemens PR, Hoffman EP. Exon-Skipping in duchenne muscular dystrophy. J Neuromuscul Dis. 2021. https://doi.org/10.3233/JND-210682.
Verdonschot JAJ, Hazebroek MR, Derks KWJ, et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J. 2018. https://doi.org/10.1093/eurheartj/ehx808.
Funding
The authors receive funding support from the Victor Chang Cardiac Research Institute, NSW Health, Australian Genomics, National Medical Research Council, and the Heart Foundation of Australia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Myocardial Disease
Rights and permissions
About this article

Cite this article
Santiago, C.F., Huttner, I.G. & Fatkin, D. Titin-related Cardiomyopathy: Is it a Distinct Disease?. Curr Cardiol Rep 24, 1069–1075 (2022). https://doi.org/10.1007/s11886-022-01726-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-022-01726-0