
Abstract
Purpose of Review
While morbidity and mortality remain high for amyloid cardiomyopathy (AC), increased awareness, earlier diagnosis, and advances in treatment have improved patient outcomes. This review will discuss the pathophysiology, contemporary diagnostic strategies, and novel and investigational therapeutic strategies for light-chain (AL) and transthyretin (ATTR) AC.
Recent Findings
Diagnostic strategies for AC now include cardiac magnetic resonance imaging and bone scintigraphy. Proteosome inhibitor therapy is now front-line therapy for AL AC followed by autologous stem cell transplantation. Emerging disease-modifying strategies for ATTR AC include the recently FDA-approved TTR-stabilizer, tafamadis. ATTR gene-silencing therapy and amyloid fibril degradation therapy are two other strategies under investigation. Heart transplantation and durable mechanical circulatory support remain a final potential option; however, contemporary outcomes are improving with better patient selection.
Summary
Patient outcomes for AC are expected to improve as increased awareness leads to earlier diagnosis and prompt treatment with emerging pharmacotherapy or advanced heart therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyloidosis is a multisystem disease caused by the deposition of misfolded insoluble fibrillar proteins into tissues leading to organ dysfunction, including within the heart [1]. The most common types of amyloidosis which affect the heart (amyloid cardiomyopathy) are light chain (AL) and transthyretin (ATTR), accounting for 95% of all cases [2, 3]. We will review the pathophysiology and clinical syndrome of amyloid cardiomyopathy (AC) as well as contemporary diagnostic strategies including the use of advanced imaging and biomarkers. Then, we will discuss novel therapeutic agents approved, and under investigation, that target the pathogenesis of disease on multiple levels.
Pathophysiology
Several proteins have been identified to potentially lead to AC (Table 1). The most commonly encountered abnormal proteins are due to light chain (AL) due to plasma cell dyscrasia in the bone marrow and transthyretin, a transport protein produced by the liver. Amyloidosis due to transthyretin typically occurs either as wild-type (normal) transthyretin (ATTRwt) or mutant transthyretin (ATTRm) protein sequence [5]. More than 120 mutations have been reported that lead to ATTRm with certain mutations having a propensity to lead to a predominant cardiac phenotype of AC and others causing a predominantly neuropathic phenotype. ATTRwt (previously known as senile systemic amyloidosis) typically affects older male patients. ATTRm generally presents at an earlier age and is inherited in an autosomal dominant trait. Transthyretin is a 127 amino acid protein which circulates as a tetramer and carries the thyroid hormone thyroxine and retinol-binding protein bound to retinol. The pathogenicity of ATTR is related to the instability of the tetramers which exist in equilibrium with its constituent monomers. The monomers misfold to form prefibrillar proteins which then aggregate to form amyloid fibrils which deposit in various organs with a particular predilection for, but not limited to, the heart and nervous system. There are also data to suggest that the pre-fibrillar protein aggregates may also directly contribute to organ toxicity [6, 7].
In cases of AL cardiomyopathy, clonal production of immunoglobulin (Ig) light chain proteins by bone marrow leads to amyloidogenic deposition within organ beds. The mechanisms of injury are not entirely understood. Light chain proteins seem to provoke direct oxidative stress as well as apoptosis in cardiomyocytes [8]. The deposition of misfolded proteins leads to a stiff, non-compliant myocardium, and the extracellular space is expanded by deposits, causing myocardial hypertrophy and restrictive cardiomyopathy with diastolic abnormalities observed on echocardiography [9,10,11].
Clinical Presentation and Diagnostic Workup
AL amyloidosis is associated with a spectrum of plasma cell disorders, including multiple myeloma, B cell lymphoma, and Waldenström macroglobulinemia [3]. The light chains often deposit within the kidneys and the heart, leading to disruption of cellular integrity, and possibly contribute to cell injury and death. Cardiac involvement is the second most common involvement after renal involvement which typically presents as nephrotic syndrome. An early diagnosis of AL cardiac amyloidosis is critical as the median survival of untreated symptomatic disease is approximately 6 months [12]. The clinical manifestations of cardiac AL amyloidosis include exertional shortness of breath, edema, and ascites. Syncope is associated with a poor prognosis [13]. The physical examination in AL amyloidosis is similar to patients with heart failure, with possible additional findings of macroglossia, periorbital bruising, and numbness [14]. The electrocardiogram (ECG) may show low voltage out of proportion to the degree of hypertrophy seen on echocardiography [15]. Low-voltage QRS complexes are more common in AL amyloidosis compared to ATTR. Other electrocardiographic findings may include the presence of a pseudoinfarct pattern in inferior and precordial leads, evidence of conduction block or atrial fibrillation. The echocardiogram demonstrates concentric left ventricular hypertrophy. Diastolic assessment virtually always demonstrates reduced tissue doppler velocity as well as diminutive transmitral A-wave on pulse wave doppler. Global longitudinal strain demonstrates near normal longitudinal strain of the apex [16]. Other findings on echocardiography may include pericardial effusion [17, 18] or asymmetric septal thickening with an outflow tract gradient [11, 19]. The right ventricle may also be hypertrophied and the atrial are typically enlarged. Cardiac magnetic resonance (CMR) may demonstrate difficulty in nulling the myocardium following gadolinium injection and subendocardial delayed enhancement [20, 21]. These findings are not specific to amyloid cardiomyopathy, but raise the suspicion. In comparing cases of AL to ATTR cardiac amyloidosis as assessed by CMR, left ventricular mass is lower in AL while late gadolinium enhancement (LGE) is less extensive; however, the type of AC cannot be usually distinguished on CMR [22]. Biochemical clues to the diagnosis of AC include elevated NT-proBNP possibly due to direct myocyte compression by amyloid fibrils and mildly but persistently elevated serum troponin [23]. The extent of elevation of these biomarkers has prognostic significance in AL [23, 24] and ATTR-wt [25] AC. An abnormal serum-free light chain kappa/lambda ratio is commonly found in AL and serum or urine immunofixation is the preferred test over protein electrophoresis for assessment of a monoclonal band. Lastly, the definitive diagnosis can be made with Congo Red staining of an endomyocardial biopsy and use of proteomic evaluation by mass spectrometry to determine amyloid subtyping. The sensitivity of endomyocardial biopsy for the diagnosis of AC is almost 100%. Complications of endomyocardial biopsy are low (1–2%), but include transient dysrhythmias, bleeding, and rarely, perforation or tricuspid valve injury.
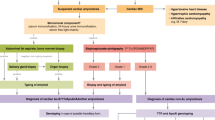
ATTR-wt cardiomyopathy more frequently affects men (90%). In the USA, the most common mutations of ATTR-m leading to AC include T60A, V30M, and V122I [27,28,29]. V122I is the most commonly seen mutation carried by 3–4% of the African American population and the presence of this mutation increases the risk for clinical heart failure. Beyond symptoms and signs of heart failure, clues for ATTR amyloidosis include carpal tunnel syndrome [30,31,32,33], lumbar spinal stenosis [31], biceps tendon rupture, peripheral, or autonomic neuropathy. Aortic stenosis is a common association with ATTR with 16% of patients undergoing transaortic valve replacement having evidence of AC [34]. Laboratory testing should demonstrate normal serum immunofixation and immunoglobulin light chain-free assay (excluding AL amyloidosis) and rarely urinalysis demonstrating nephrotic range proteinuria. ECG and echocardiographic findings are similar between AL and ATTR amyloidosis. Within the ATTR subclasses, ATTRwt has shown greater left ventricular wall thickness and lower ejection fraction as compared to ATTRm [35]. CMR is a useful tool in the diagnosis of ATTR cardiomyopathy with findings including LGE over the entire subendocardial circumference. Beyond CMR, the use of radionuclide imaging has found an important role in the evaluation of suspected ATTR cardiomyopathy. Radionuclide bone scintigraphy with technetium-labeled bisphosphonates (e.g., pyrophosphate) can demonstrate localized cardiac amyloid deposits. In an analysis of 1217 patients with suspected cardiac amyloidosis, myocardial radiotracer uptake on bone scintigraphy was greater than 99% sensitive and 86% specific for ATTR amyloidosis [36]. Grade 2 or 3 myocardial radiotracer uptake (i.e., equal to or greater than uptake in ribs) on bone scintigraphy and the absence of free light chains in the serum and monoclonal protein in serum or urine is essentially diagnostic of ATTR cardiomyopathy. It is essential to rule out AL amyloidosis when relying solely on bone scintigraphy for the diagnosis of ATTR cardiomyopathy as rarely AL cardiomyopathy may present with increased myocardial uptake on bone scintigraphy. Beyond imaging, detectable serum troponin I and T is less often seen in ATTR cardiomyopathy as compared to AL, while natriuretic peptides are often elevated in confirmed cases [37]. For ATTR-wt cardiomyopathy, both these biomarkers have prognostic significance [25]. A potential approach to biopsy in suspected AC cases could include verification of deposits in either fat, bone marrow, lip, skin, salivary gland, or gastrointestinal tract. However, the sensitivity of these tests to diagnose amyloidosis is variable. Therefore, if this biopsy is negative, evaluation of heart, nerve, or kidney biopsy can be considered. Lastly, mass spectroscopy is preferred over immunohistochemical/immunogold staining with electron microscopy after tissue is obtained. An algorithm (Fig. 1) for the diagnosis of AC has been proposed by Nativi-Nicolau [38].

A proposed algorithm for the diagnosis of amyloid cardiomyopathy. ECG, electrocardiogram; Echo, echocardiogram; AL, light chain amyloidosis; ATTR, amyloid transthyretin; MRI, magnetic resonance imaging; ATTRm, hereditary transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis (from: Nativi-Nicolau J, Maurer MS. Curr Opin Cardiol. 2018;33:571–9, with permission from Wolters Kluwer Health, Inc.) [38]
Prognosis
The prognosis of patients with either AL or ATTR cardiomyopathy depends on the extent and severity of organ involvement, although overall, untreated AL has a much more rapidly progressive course.
AL Amyloidosis
Kumar et al. [23] reported on an updated prognostic staging system for patients with cardiac AL amyloidosis identifying three independent predictors of mortality: (1) the difference between involved and uninvolved light chains (free light chain difference: ≥ 18 mg/dL), (2) cardiac troponin T (cTnT: ≥ 0.025 ng/mL), and (3) NT-proBNP ≥ 1800 pg/mL. Overall survival significantly decreased for each additional biomarker above the cutoff values: 94.1 months (0 points), 40.3 months (1 point), 14.0 months (2 points), and 5.8 months (3 points). This scoring system predates the use of proteosome inhibitors such as bortezomib and therefore limits its contemporary applicability. An earlier prognostic tool utilized only cTnT and NT-proBNP for risk stratification [39]. Beyond the use of biomarkers, the degree of LGE seen on CMR has prognostic utility for long-term mortality in both AL and ATTR [40].
ATTR
Median survival in a single-center study of patients with ATTRwt was 3.6 years with outcomes remaining static over the last 30–40 years [25]. The authors report on three stages of disease based on whether cardiac biomarker values were above or below an NT-proBNP cutoff of 3000 pg/mL and troponin T cutoff of 0.05 ng/mL. Four-year overall survival worsened with increasing stage with stage I (both values below cutoff), stage II (one value above cutoff), and stage III both values above cutoff) having 4-year overall survival of 57%, 42%, and 18% (p < 0.001), respectively. For ATTRm, untreated survival ranges between 2 and 15 years and dependent upon mutation sub-type [27]. The V122I or valine to isoleucine mutation is the most common sub-type in the US and carries an 80% survival at 1 year and 28% at 5 years, respectively [27]. Updated scoring systems and prognostication models will be necessary for AL and ATTR as novel therapies bend survival curves.
Medical Management
Medical management of amyloid cardiomyopathy is directed at reducing congestion, minimizing symptomatic hypotension, and controlling cardiac arrhythmias. To that end, loop and thiazide diuretics with or without adjunctive use of mineralocorticoid receptor antagonists are preferred agents for targeting congestion. Diuretics with high bioavailability may be needed due to poor absorption from congested bowel. Avoidance of beta-blocker and calcium-channel blocker therapy as well as afterload-reducing agents is often necessary due to reliance on heart rate and inotropy to maintain an adequate cardiac output, while also avoiding hypotension. Amyloid patients may be particularly prone to bradyarrhythmic effects of non-dihydropyridine calcium channel blockers [41,42,43]. The negative inotropic effects may be profound, possibly because of avid binding of drug to amyloid fibrils and may depress compensatory heart rate responses to low stroke volume and cardiac output [44]. Symptomatic hypotension may be tempered with compression stockings or midodrine therapy.
Rhythm Management
Rhythm and conduction abnormalities in patients with cardiac amyloidosis are not uncommon. In patients presenting with symptomatic atrioventricular (AV) block, pacemaker implantation may be required [45, 46]. Arrhythmic death is common in the amyloid cardiomyopathy population [47], yet the role of implantable cardioverter-defibrillator (ICD) remains unclear and appropriate patient selection is a challenging endeavor [47,48,49,50]. The Stanford Amyloid Center’s ICD implantation criteria may assist with patient selection [47]. Atrial arrhythmias are common and management with direct current cardioversion (DCCV) is frequently required. Caution should be taken as patients with AC undergoing DCCV have high rates of cancelation due to identification of intracardiac thrombus formation despite adequate anti-coagulation [51].
Novel Therapeutic Strategies
The last decade has brought with it significant changes in the management options for disease-targeting strategies that modify the course of AC on the molecular level. ATTR therapy can be divided into three categories: (1) TTR silencers—therapies that prevent production of abnormal ATTR; (2) TTR stabilizers that prevent monomer dissociation and therefore aberrant protein folding; and (3) TTR fibril disruptors that lead to the removal of pathogenic fibrils from cardiac and other tissue. We will discuss these therapeutic strategies further below. AL amyloidosis on the other hand targets plasma cell production and thus the eventual production of amyloidogenic protein; therapy for AL amyloidosis is primarily managed by hematologists, and therefore, we will only provide an abbreviated discussion on therapeutic options. We refer the reader to more comprehensive reviews on AL amyloidosis [52, 53].
ATTR Amyloidosis
TTR Silencers
The anti-sense oligonucleotide, inotersen, was studied in the randomized, double-blind, placebo-controlled NEURO-TTR trial [54] in patients with hereditary or familial TTR and polyneuropathy. There was a significant improvement in neurologic outcomes favoring the inotersen-treated group. Sixty-three percent of the study population had cardiomyopathy (defined as interventricular wall thickness 13 mm or greater in the absence of hypertension). Despite randomization, the presence of cardiomyopathy was higher in the inotersen group compared to the placebo group. In the subset of patients with cardiomyopathy, there was no significant difference in global longitudinal strain or other echocardiographic variables following 15 months of treatment. Side effects of this therapy included a higher rate of glomerulonephritis and thrombocytopenia than what was seen in the placebo group. Currently, this therapy is approved for use in the USA, Europe, and Canada with an indication for stage 1 or stage 2 polyneuropathy in patients with hereditary ATTR. A 24-month, phase 2, open-label study of 50 patients with cardiac amyloidosis due to both ATTRm and ATTRwt is currently listed on the clinicaltrials.gov website [26]. To date, no dedicated studies evaluating inotersen efficacy in cardiac amyloidosis have been completed.
Patisiran is a novel therapy that limits TTR production utilizing RNA interference (RNAi) or small-interfering RNAs (siRNA) to target hepatocyte-derived mRNA and cause cleavage before protein translation can occur. The APOLLO Trial [55] was a phase 3, randomized, placebo-controlled trial of intravenous patisiran infusions in patients with hereditary TTR and polyneuropathy. Patisiran therapy resulted in a significant improvement in neurologic outcomes as well as an increase in gait speed, quality of life, and ability to complete activities of daily living. The major adverse event was mild or moderate infusion-related reactions with a similar overall incidence of adverse events in placebo and treatment groups. Exploratory cardiovascular endpoints from a post hoc analysis [56•] included echocardiographic, hemodynamic, and cardiac biomarker evaluation in addition to clinical outcomes in 126 patients (56% of initial APOLLO cohort). Mean LV wall thickness, global longitudinal strain, NT-proBNP, cardiac output, and end-diastolic volume all significantly improved in the treatment groups at 18 months compared to placebo. There was a trend towards a reduction in cardiac hospitalizations and all-cause mortality, but did not reach statistical significance. The incidence of cardiac arrhythmias was lower in the patisiran treated versus placebo group (19% vs 29%). Similar to inotersen, patisiran was recently FDA-approved for use in the USA in patients with polyneuropathy due to hereditary TTR.
TTR Stabilizers
Tafamidis is a novel oral therapy that binds to the thyroid-binding site of the TTR tetramer and inhibits dissociation into monomers thereby decreasing fibril aggregates and tissue deposition of protein. In an international, multicenter, double-blind, placebo-controlled phase 3 trial of patients with both hereditary and wild-type TTR cardiomyopathy, tafamidis was found to be associated with lower all-cause mortality, fewer cardiovascular-related hospitalizations, and a slower decline in quality of life compared to placebo [57•]. Decrease in 6-min walk test was attenuated in the pooled tafamidis cohort. Favorable outcomes were limited to patients with NYHA functional I-II, suggesting the importance of early diagnosis and prompt therapy initiation prior to target organ damage occurring. Tafamadis was FDA-approved on May 3rd, 2019 for amyloid cardiomyopathy [58]. Evaluation of the long-term safety of tafamidis is ongoing [59].
Like tafamidis, AG10 is an orally administered TTR stabilizer. The results of a recent phase 2 trial [60] in patients with ATTR cardiac amyloidosis and NYHA functional class II-III symptoms demonstrated AG10 as tolerable and able to restore serum TTR levels to a normal range. The phase 3 trial of AG10 in (ATTRibute-CM; NCT03860935) is underway with an estimated completion in November, 2022.
Diflunisal is a nonsteroidal anti-inflammatory agent that has been evaluated for efficacy as a TTR stabilizer with prior demonstration of improvement in neurological outcomes [61]. To date, there are no completed or ongoing randomized studies evaluating efficacy of diflunisal in cardiac patients; additionally, adverse events including renal dysfunction, gastrointestinal bleeding, and fluid retention make this a challenging therapy to pursue further in an already at-risk, heart failure population.
Fibril Disruptors
Doxycycline, an antibiotic and tetracycline derivative, has been shown to disrupt amyloid fibrils in vitro [62] and in mouse models [63]. Taursodeoxycholic acid (TUDCA), a bile acid used in the treatment of cholelithiasis, decreases toxic TTR aggregates in mice [64]. Combined use of doxycycline and TUDCA in transgenic mice appears to synergistically lower TTR deposition [65]. A phase II, open-label study of the combination of doxycycline/TUDCA in 20 patients with ATTR demonstrated stability of cardiac and neurologic status over time with an acceptable toxicity profile [66]. A study of serial echocardiographic assessment of left ventricular function over 18 months in patients with TTR cardiomyopathy treated with doxycycline/TUDCA has been completed, but the results have not yet been reported [67]. The study findings will further inform the role of this therapeutic strategy.
In vitro evaluation of epigallocathechin-3-gallate (EGCG) or green tea extract has been shown to bind to amyloidogenic light chains and prevent the subsequent formation of amyloid fibrils [68]. A study of seven patients following 12 months of daily EGCG consumption found a reduction in LV mass as measured by cardiac MRI [69]. In a similar fashion, an observational study of EGCG in 59 patients with cardiac AL amyloidosis identified improvement in NYHA functional class and LVEF [70].
A phase 1 trial of 15 patients with systemic amyloidosis evaluated the combination therapy of (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) and anti-serum amyloid protein (SAP) antibody [71]. This strategy relies on the binding of CPHPC to circulating SAP with subsequent anti-SAP antibody binding to tissue SAP. This leads to eventual complement activation and then clearance of amyloid protein by macrophages. SAP scintigraphy and MRI demonstrated reduction of tissue amyloid as measured by SAP scintigraphy and MRI [71•, 72•]. This combination therapy had no serious adverse events and was well-tolerated.
AL Amyloidosis
Chemotherapeutic strategies leading to destruction of plasma cells, and thus the eventual production of amyloid fibrils, are the cornerstone of AC treatment with the addition of autologous hematopoietic stem cell transplantation (HSCT) reserved for select patient cases [52, 73]. The oldest treatment regimen includes combination melphalan and steroid therapy [52, 53]. With the FDA approval of proteosome inhibitors, first-line combination therapy now includes bortezomib, dexamethasone, and cyclophosphamide with high rates of hematologic and organ response [74,75,76,77]. Earlier identification of disease with prompt chemotherapeutic intervention using either melphalan-based or bortezomib-based has been associated with better outcomes than more advanced disease [76, 78]. Ixazomib is an oral proteasome inhibitor currently in phase 3 trial evaluating 2-year cardiac deterioration rates in patients with relapsed or refractory AL amyloidosis [79]. Carfilzomib, another proteosome inhibitor, is also under study [80].
The anti-CD38 monoclonal antibody, daratumumab, is another option that has demonstrated reasonable hematologic and complete response rates [81, 82]. This therapy is fairly well-tolerated, with only minor infusion-related reactions observed. There is an ongoing phase I-II trial of daratumumab in patients with AL amyloidosis and cardiac involvement with an expected completion date of August 2020 [83].
Lenalidomide is a thalidomide derivative and immunomodulator with use in the treatment of multiple myeloma, however has also demonstrated efficacy in patients with AL amyloidosis and cardiac involvement [84, 85]. Pomalidomide, a newer generation immunomodulatory agent, may also have a role for use in patients with cardiac AL amyloidosis [86,87,88].
Although these therapies appear to be very effective at reducing light chain burden in AL AC, their impact on progression of cardiac disease has been difficult to assess and select patients may need to be considered for advanced therapies for heart failure.
Heart Replacement Therapy Strategies
Mechanical Circulatory Support
The small LV cavity size and restrictive nature of cardiac amyloidosis generally make left ventricular assist device therapy a suboptimal strategy for durable mechanical support [89,90,91]. With durable LVAD therapy, a small LV cavity may increase the risk of cannula obstruction, suction events, suboptimal flow profiles, and pump thrombosis [89]. Additionally, right ventricular dysfunction is often present in these patients and these patients may be at increased risk for RV failure after durable LVAD implantation [89]. Patients with larger LV end diastolic dimension above 46 mm have better outcomes with durable LVAD [91]. Temporary devices such as the Impella (Abiomed, CA, USA) may present challenges to appropriate positioning. With this in mind, there are reports of success with the total artificial heart (Syncardia, Tucson, AZ, USA) as a bridge to heart transplantation strategy [92, 93].
Heart Transplantation
Historically, post-heart transplantation survival for patients with amyloid cardiomyopathy has been worse than that of non-amyloid cardiomyopathy [94,95,96]. Waitlist mortality for patients with CA is greater than those with dilated cardiomyopathies despite being listed at a lower acuity status [96]. Contemporary data suggest that post-transplant outcomes are now approaching that of non-amyloid cardiomyopathy [95, 97•]. This shift in outcomes is likely due to a standardized approach to patient selection [98], which includes a comprehensive evaluation for clinically significant extra-cardiac involvement of disease. Likely in response to stricter scrutiny, a higher percentage of ATTR cardiomyopathy patients are now undergoing heart transplantation as compared to AL amyloid cardiomyopathy [95]. Nonetheless, novel treatment options in patients with AL amyloid cardiomyopathy, as discussed above, have certainly contributed to improved post-heart transplantation survival particularly when combined with autologous stem cell transplant performed after heart transplantation [99•]. Complete hematologic response to chemotherapy in AL amyloid patients post-HT is associated with good long-term survival [100].
Conclusions
The past decade has brought with it significant improvements in survival in patients with cardiac amyloidosis due to AL, ATTRm, and ATTRwt. Early diagnosis, prompt initiation of therapy when indicated, and improvements in patient selection through appropriate evaluation for extra-cardiac manifestations of disease all have contributed to improvements in post-HT outcomes. The disease-modifying therapies of ATTR will likely take greater hold in the management of patients with cardiac involvement as the results of ongoing clinical trials become available. The role for combination ATTR therapy is enticing as it would target multiple stages of pathogenesis. The future of amyloid therapy appears bright.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130:88–98.
Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126:1286–300.
Buxbaum JN, Chuba JV, Hellman GC, Solomon A, Gallo GR. Monoclonal immunoglobulin deposition disease: light chain and light and heavy chain deposition diseases and their relation to light chain amyloidosis. Clinical features, immunopathology, and molecular analysis. Ann Intern Med. 1990;112:455–64.
Falk RH, Alexander KM, Liao R, Dorbala S. AL (light-chain) cardiac amyloidosis. J Am Coll Cardiol. 2016;68:1323–41.
Sipe JD, Benson MD, Buxbaum JN, Ikeda S-I, Merlini G, Saraiva MJM, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 nomenclature guidelines. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2016;23:209–13.
Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ. Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci. 2001;21:7576–86.
Mendes Sousa M, Cardoso I, Fernandes R, Guimarães A, Saraiva MJ. Deposition of transthyretin in early stages of familial Amyloidotic polyneuropathy. Am J Pathol. 2001;159:1993–2000.
Shi J, Guan J, Jiang B, Brenner DA, del Monte F, Ward JE, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38α MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–93.
Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–76.
Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J. 2005;32:178–84.
Falk RH, Quarta CC. Echocardiography in cardiac amyloidosis. Heart Fail Rev. 2015;20:125–31.
Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817–22.
Chamarthi B, Dubrey SW, Cha K, Skinner M, Falk RH. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80:1242–5.
Baker KR, Rice L. The Amyloidoses: clinical features, diagnosis and treatment. Methodist DeBakey Cardiovasc J. 2012;8:3–7.
Mussinelli R, Salinaro F, Alogna A, Boldrini M, Raimondi A, Musca F, et al. Diagnostic and prognostic value of low QRS voltages in cardiac AL amyloidosis. Ann Noninvasive Electrocardiol Off J Int Soc Holter Noninvasive Electrocardiol Inc. 2013;18:271–80.
Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart Br Card Soc. 2012;98:1442–8.
Pericardial tamponade, a new complication of amyloid heart disease - The American Journal of Medicine [Internet]. [cited 2019 Apr 29]. Available from: https://www.amjmed.com/article/0002-9343(82)90939-1/pdf.
Navarro JF, Rivera M, Ortuño J. Cardiac tamponade as presentation of systemic amyloidosis. Int J Cardiol. 1992;36:107–8.
Philippakis AA, Falk RH. Cardiac amyloidosis mimicking hypertrophic cardiomyopathy with obstruction: treatment with disopyramide. Circulation. 2012;125:1821–4.
Kwong RY, Heydari B, Abbasi S, Steel K, Al-Mallah M, Wu H, et al. Characterization of cardiac amyloidosis by atrial late gadolinium enhancement using contrast-enhanced cardiac magnetic resonance imaging and correlation with left atrial conduit and contractile function. Am J Cardiol. 2015;116:622–9.
Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20:133–44.
Dungu JN, Valencia O, Pinney JH, Gibbs SDJ, Rowczenio D, Gilbertson JA, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:133–42.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–95.
Kumar SK, Gertz MA, Dispenzieri A. Validation of Mayo Clinic staging system for light chain amyloidosis with high-sensitivity troponin. J Clin Oncol. 2018;37:171–3.
Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68:1014–20.
24 Month Open Label Study of the Tolerability and Efficacy of Inotersen in TTR Amyloid Cardiomyopathy Patients - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 Apr 24]. Available from: https://clinicaltrials.gov/ct2/show/NCT03702829.
Swiecicki PL, Zhen DB, Mauermann ML, Kyle RA, Zeldenrust SR, Grogan M, et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid. 2015;22:123–31.
Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372:21–9.
Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20:163–78.
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–91.
Aus dem Siepen F, Hein S, Prestel S, Baumgärtner C, Schönland S, Hegenbart U, et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol Off J Ger Card Soc. 2019.
Maurer MS, Ruberg FL. Early diagnosis of cardiac amyloidosis by carpal tunnel surgery: is it all in the wrist? J Am Coll Cardiol. 2018;72:2051–3.
Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72:2040–50.
Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–87.
Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129:1840–9.
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12.
Damy T, Jaccard A, Guellich A, Lavergne D, Galat A, Deux J-F, et al. Identification of prognostic markers in transthyretin and AL cardiac amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2016;23:194–202.
Nativi-Nicolau J, Maurer MS. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018;33:571–9.
Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751–7.
Marianna F, Silvia P, Patricia R, Amna A-G. Treibel Thomas a., Banypersad Sanjay M., et al. prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132:1570–9.
Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55:1645.
Griffiths BE, Hughes P, Dowdle R, Stephens MR. Cardiac amyloidosis with asymmetrical septal hypertrophy and deterioration after nifedipine. Thorax. 1982;37:711–2.
Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104:618–20.
Gertz MA, Skinner M, Connors LH, Falk RH, Cohen AS, Kyle RA. Selective binding of nifedipine to amyloid fibrils. Am J Cardiol. 1985;55:1646.
Algalarrondo V, Dinanian S, Juin C, Chemla D, Bennani SL, Sebag C, et al. Prophylactic pacemaker implantation in familial amyloid polyneuropathy. Heart Rhythm. 2012;9:1069–75.
González-López E, Gagliardi C, Dominguez F, Quarta CC, de Haro-del Moral FJ, Milandri A, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017;38:1895–904.
Varr BC, Zarafshar S, Coakley T, Liedtke M, Lafayette RA, Arai S, et al. Implantable cardioverter-defibrillator placement in patients with cardiac amyloidosis. Heart Rhythm. 2014;11:158–62.
Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the Management of Patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–867.
Lin G, Dispenzieri A, Kyle R, Grogan M, Brady PA. Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol. 2013;24:793–8.
Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, Sack F-U, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm. 2008;5:235–40.
El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, et al. Direct current cardioversion of atrial arrhythmias in adults with cardiac amyloidosis. J Am Coll Cardiol. 2019;73:589–97.
Alexander KM, Singh A, Falk RH. Novel pharmacotherapies for cardiac amyloidosis. Pharmacol Ther. 2017;180:129–38.
Kastritis E, Dimopoulos MA. Recent advances in the management of AL amyloidosis. Br J Haematol. 2016;172:170–86.
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med [Internet]. 2018 [cited 2019 Apr 24]; Available from: https://www.nejm.org/doi/10.1056/NEJMoa1716793?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov.
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21.
• Solomon SD, David A, Arnt K, Martha G, Alejandra G-D, Maurer Mathew S, et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139:431–43. This study reported on the significant improvement in several exploratory cardiovascular endpoints.
• Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J med [internet]. 2018 [cited 2019 Apr 16]; available from: https://www.nejm.org/doi/10.1056/NEJMoa1805689?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov. This study lead to the eventual FDA-approval of the drug tafamadis for amyloid cardiomyopathy.
Commissioner O of the. FDA approves new treatments for heart disease caused by a serious rare disease, transthyretin mediated amyloidosis [Internet]. FDA. 2019 [cited 2019 May 6]. Available from: /news-events/press-announcements/fda-approves-new-treatments-heart-disease-caused-serious-rare-disease-transthyretin-mediated.
Long-term Safety of Tafamidis in Subjects With Transthyretin Cardiomyopathy - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 Apr 29]. Available from: https://clinicaltrials.gov/ct2/show/NCT02791230.
Judge DP, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74:285–95.
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–67.
Cardoso I, Merlini G, Saraiva MJ. 4′-iodo-4′-Deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: screening for TTR fibril disrupters. FASEB J. 2003;17:803–9.
Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20:234–9.
Macedo B, Batista AR, Ferreira N, Almeida MR, Saraiva MJ. Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of familial Amyloidotic polyneuropathy. Biochim Biophys Acta. 1782;2008:517–22.
Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74.
Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19:34–6.
Tolerability and Efficacy of a Combination of Doxycycline and TUDCA in Patients With Transthyretin Amyloid Cardiomyopathy - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 Apr 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT01855360.
Hora M, Carballo-Pacheco M, Weber B, Morris VK, Wittkopf A, Buchner J, et al. Epigallocatechin-3-gallate preferentially induces aggregation of amyloidogenic immunoglobulin light chains. Sci Rep. 2017;7:41515.
aus dem Siepen F, Buss SJ, Andre F, Seitz S, Giannitsis E, Steen H, et al. Extracellular remodeling in patients with wild-type amyloidosis consuming epigallocatechin-3-gallate: preliminary results of T1 mapping by cardiac magnetic resonance imaging in a small single center study. Clin Res Cardiol Off J Ger Card Soc. 2015;104:640–7.
Mereles D, Buss SJ, Hardt SE, Hunstein W, Katus HA. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin Res Cardiol Off J Ger Card Soc. 2010;99:483–90.
• Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373:1106–14.
• Richards DB, Cookson LM, Barton SV, Liefaard L, Lane T, Hutt DF, et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med. 2018;10:eaan3128. This study is notable because it provides additional proof-of-concept for removal of amyloid deposition from tissues; whether this improves outcomes in amyloid cardiomyopathy is not yet clear.
Falk RH, Alexander KM, Liao R, Dorbala S. AL (light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68:1323–41.
Huang X, Wang Q, Chen W, Ren G, Liu Z. Bortezomib with dexamethasone as first-line treatment for AL amyloidosis with renal involvement. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2016;23:51–7.
Milani P, Gertz MA, Merlini G, Dispenzieri A. Attitudes about when and how to treat patients with AL amyloidosis: an international survey. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2017;24:213–6.
Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126:612–5.
Venner CP, Lane T, Foard D, Rannigan L, Gibbs SDJ, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119:4387–90.
Landau H, Hassoun H, Rosenzweig MA, Maurer M, Liu J, Flombaum C, et al. Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia. 2013;27:823–8.
Study of Dexamethasone Plus IXAZOMIB (MLN9708) or Physicians Choice of Treatment in Relapsed or Refractory Systemic Light Chain (AL) Amyloidosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 May 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT01659658.
A Safety Study of Carfilzomib in Patients With Previously-Treated Systemic Light Chain Amyloidosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 May 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT01789242.
Kaufman GP, Schrier SL, Lafayette RA, Arai S, Witteles RM, Liedtke M. Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis. Blood. 2017;130:900–2.
Sher T, Fenton B, Akhtar A, Gertz MA. First report of safety and efficacy of daratumumab in 2 cases of advanced immunoglobulin light chain amyloidosis. Blood. 2016;128:1987–9.
Daratumumab for the Treatment of Patients With AL Amyloidosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2019 May 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT02841033.
Kumar SK, Hayman SR, Buadi FK, Roy V, Lacy MQ, Gertz MA, et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood. 2012;119:4860–7.
Mahmood S, Venner CP, Sachchithanantham S, Lane T, Rannigan L, Foard D, et al. Lenalidomide and dexamethasone for systemic AL amyloidosis following prior treatment with thalidomide or bortezomib regimens. Br J Haematol. 2014;166:842–8.
Dispenzieri A, Buadi F, Laumann K, LaPlant B, Hayman SR, Kumar SK, et al. Activity of pomalidomide in patients with immunoglobulin light-chain amyloidosis. Blood. 2012;119:5397–404.
Sanchorawala V, Shelton AC, Lo S, Varga C, Sloan JM, Seldin DC. Pomalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 1 and 2 trial. Blood. 2016;128:1059–62.
Sharpley FA, Manwani R, Mahmood S, Sachchithanantham S, Lachmann H, Gilmore J, et al. Real world outcomes of pomalidomide for treatment of relapsed light chain amyloidosis. Br J Haematol. 2018;183:557–63.
Yan T, Pereira Naveen L, Shah Dipesh K, Barry B, Schirger John A, Kushwaha Sudhir S, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail. 2011;4:266–75.
Swiecicki PL, Edwards BS, Kushwaha SS, Dispenzieri A, Park SJ, Gertz MA. Left ventricular device implantation for advanced cardiac amyloidosis. J Heart Lung Transplant. 2013;32:563–8.
Grupper A, Park SJ, Pereira NL, Schettle SD, Gerber Y, Topilsky Y, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: improving outcomes for a lethal disease. J Heart Lung Transplant. 2015;34:1042–9.
Scully MS, Wessman DE, McKee JM, Francisco GM, Nayak KR, Kobashigawa JA. Total artificial heart implantation as a bridge to heart transplantation in an active duty service member with amyloid cardiomyopathy. Mil Med. 2017;182:e1858–60.
Gerosa G, Scuri S, Iop L, Torregrossa G. Present and future perspectives on total artificial hearts. Ann Cardiothorac Surg. 2014;3:595–602–602.
DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant Off Publ Int Soc Heart Transplant. 2012;31:1269–75.
Davis MK, Lee PHU, Witteles RM. Changing outcomes after heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2015;34:658–66.
Panhwar MS, Al-Kindi SG, Tofovic D, Oliveira GH, Ginwalla M. Waitlist Mortality of Amyloid Cardiomyopathy Patients Listed for Heart Transplantation and Implications for Organ Allocation. J Card Fail [Internet]. 2019 [cited 2019 Apr 27]; Available from: http://www.sciencedirect.com/science/article/pii/S107191641930421X.
• Kristen AV, Kreusser MM, Blum P, Schönland SO, Frankenstein L, Dösch AO, et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. J Heart Lung Transplant. 2018;37:611–8. This provides important evidence for improving outcomes in select patients undergoing heart transplantation for amyloid cardiomyopathy.
Varr BC, Liedtke M, Arai S, Lafayette RA, Schrier SL, Witteles RM. Heart transplantation and cardiac amyloidosis: approach to screening and novel management strategies. J Heart Lung Transplant. 2012;31:325–31.
• Trachtenberg BH, Kamble RT, Rice L, Araujo-Gutierrez R, Bhimaraj A, Guha A, et al. Delayed autologous stem cell transplantation following cardiac transplantation experience in patients with cardiac amyloidosis. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2019.
Grogan M, Gertz M, McCurdy A, Roeker L, Kyle R, Kushwaha S, et al. Long term outcomes of cardiac transplant for immunoglobulin light chain amyloidosis: the Mayo Clinic experience. World J Transplant. 2016;6:380–8.
Funding
Jignesh Patel reports grants from the National Institutes of Health (Grant number: 1UO1AI136816-01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Aaron M. Wolfson and Kevin S. Shah declare that they have no conflict of interest.
Jignesh Patel reports grants, personal fees, non-financial support and other from Alnylam Pharmaceuticals; personal fees, non-financial support and other from Akcea Pharmaceuticals; grants, personal fees, non-financial support and other from Pfizer Inc.; grants from Ionis Pharmaceuticals; grants from Alexion Pharmaceuticals; grants from CareDx Inc., and grants from Genzyme Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Heart Failure
Rights and permissions
About this article

Cite this article
Wolfson, A.M., Shah, K.S. & Patel, J.K. Amyloid and the Heart. Curr Cardiol Rep 21, 164 (2019). https://doi.org/10.1007/s11886-019-1230-9
Published:
DOI: https://doi.org/10.1007/s11886-019-1230-9