
Abstract
Increase in heart rate represents a significant contribution in the pathophysiology of coronary artery disease and heart failure, by promoting atherosclerotic process and endothelial dysfunction. Thus, it negatively influences cardiovascular risk in the general population. The aim of this review is to analyze the current, controversial, and future role of ivabradine as an anti-anginal agent in the setting of coronary artery disease without heart failure. Ivabradine represents a selective heart rate-lowering agent that increased diastolic perfusion time and improving energetics in the ischemic myocardium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In contrast to most other vascular beds, myocardial oxygen extraction is near maximal at rest. The major determinants of myocardial oxygen consumption are heart rate, systolic pressure (myocardial wall stress), and left ventricular contractility. During diastole, the coronary arterial inflow increases with a transmural gradient that favors perfusion to the sub-endocardial vessels [1]. The duration of the diastasis phase in diastole is, for the most part, regulated by the heart rate [2]; thus, the lower the heart rate, the longer the diastole, hence the time of effective myocardial perfusion. In patients with known coronary artery disease (CAD), an elevated heart rate reduces the diastolic filling time, increasing the cardiac workload, resulting in a supply–demand mismatch with subsequent ischemia and angina [3]; thus, heart rate represents a major determinant of myocardial oxygen consumption, playing a pivotal role in the pathophysiology of chronic CAD [4].
In mammalians, an elevated resting heart rate has been associated with a shorter life span [5]. In the Framingham Heart Study, individuals having higher heart rates had a progressive increase in the risk of all-cause mortality and cardiovascular mortality [6]. Also, it was seen that individuals with a higher heart rate were at a higher long‐term risk for cardiovascular events, particularly, heart failure and all‐cause death [6]. Increasing evidence has shown that an elevated heart rate is associated with a greater risk of developing hypertension and atherosclerosis, being a potent predictor of cardiovascular morbidity and mortality [3, 4].
Mechanism of Action of Ivadrabine
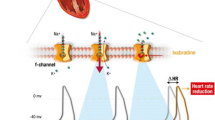
Ivabradine represents an innovative agent which acts by reducing the heart rate via a specific competitive inhibition of the so called “funny” (If) channels, a mechanism different from that of β-blockers and calcium channel blockers, two commonly prescribed anti-anginal drugs [7]. Ivabradine reduces the myocardial oxygen demand, preserving ventricular contractility, simultaneously improving oxygen supply without negative inotropic or lusitropic effects [8]. Thus, it does not decrease systemic blood pressure which is a common feature of most of the anti-anginal agents (e.g., β-blockers, calcium channel blockers, and nitrates) except for ranolazine.
Heart rate is determined by the spontaneous electrical pacemaker activity in the sinoatrial node by the If current. The “funny” (If) current channels, originally described in sinoatrial node cardiomyocytes as an inward current activated on hyperpolarization has properties appropriate for generating and modulating spontaneous pacemaker activity. Because If is controlled by intracellular cAMP and is thus activated and inhibited by β-adrenergic and muscarinic M2 receptor stimulation, respectively, it represents a basic physiological mechanism mediating autonomic regulation of heart rate [7, 8].
Ivabridine and Stable Coronary Artery Disease
The anti-anginal and anti-ischemic efficacy of ivabradine in patients with stable CAD has been tested in clinical studies comparing it either with placebo or with standard anti-anginal agents.
In a placebo-controlled, randomized trial conducted in 360 patients with stable CAD, Borer et al. [9] showed that ivabradine resulted in dose-dependent improvements in exercise tolerance parameters. At 2 weeks, ivabradine 5 and 10 mg (twice daily dose) significantly improved the time to ST-segment depression compared to placebo. Furthermore, in an open-label extension (2 to 3 months) of this trial, ivabradine reduced angina attacks from 4.14 to 0.95 times per week and decreased consumption of short acting nitrates from 2.28 to 0.50 Units per week.
The INITIATIVE study, a randomized, double-blind trial studied 939 patients comparing ivabradine directly with atenolol [10]. After 16 weeks of treatment in patients receiving ivabradine 7.5 or 10 mg twice daily vs atenolol 100 mg daily, both had similar improvements in total exercise duration at trough, as well as the number of angina attacks per week. The studay had an overall tendency to improve functional capacity and prolong time to ST-segment depression when compared to atenolol. Hence, it proved ivabradine to be non-inferior after 4 months of therapy.
There has been also studies comparing ivabradine (7.5 and 10 mg twice daily) with amlodipine 10 mg once daily in a large multicenter, l, randomized trial that included 1195 patients with stable CAD [11]. Ivabradine was non-inferior to amlodipine in improving exercise tolerance, preventing angina attacks and limiting nitrate use.
The ASSOCIATE trial evaluated the add-on ivabradine in patients already receiving atenolol in 889 patients [12]. All included patients had a positive history of symptom-limited exercise while receiving atenolol 50 mg once daily. These patients were randomized to receive either ivabradine 5 mg twice daily for 2 months, and then increased to 7.5 mg for an additional 2 months (449 patients). As an add-on therapy, ivabradine significantly increased the long-term exercise capacity, and decreased symptomatic angina.
Signify Trial and the Insignificant Role of Ivabradine
The SIGNIFY (Study Assessing the Morbidity-Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease) trial studied whether and to what extent the pharmacological reduction of an elevated resting heart rate could be associated with clinical benefits, by reducing cardiovascular events in patients with stable CAD [13••].
The SIGNIFY trial was a prospective, randomized, double-blind, multicenter trial that included 19,102 patients with documented stable CAD without heart failure followed for an average 28 months. The main objective of the trial was if the add-on therapy of ivabradine on top of conventional anti-anginal therapies was capable to reduce the primary composite end-point of death from cardiovascular causes or non-fatal myocardial infarction. Secondary end-point was defined as the primary end-point plus all-cause mortality.
There was no significant difference between the ivabradine group versus placebo group in the incidence of the primary end-point (6.8 vs 6.4 %, respectively; P = 0.20). However, in a large subgroup of patients (>12000 patients) who had class II or higher angina, ivabradine was associated with an increase in the incidence of the primary end-point compared to placebo (7.6 vs 6.5 %, P < 0.02). Moreover, the incidence of symptomatic bradycardia, asymptomatic bradycardia, QT-interval prolongation, and phosphenes were significantly greater in the ivabradine group versus placebo-treated group (P < 0.001 for all the above comparisons). There was also a trend towards more heart failure admissions in the ivabradine group (2.3 % vs 1.9 %;P = 0.07).
Interestingly, the SIGNIFY trial reported higher incidences of atrial fibrillation in the ivabradine group versus placebo group (5.3 vs 3.8 %, P < 0.001), corresponding to a 40 % increase in relative risk and 1.5 % of new cases of atrial fibrillation. It is important to emphasize that the SIGNIFY trial was considered a negative trial, by failing to demonstrate a significant reduction in cardiovascular events and cardiovascular-related mortality. In subgroup analysis of patients that reached a heart rate <60 beats/min, a J curve phenomenon of increased cardiovascular mortality, worsening angina, and increased incidence of atrial fibrillation was noted. When analyzed by different subgroups such as baseline heart rate, previous myocardial infarction, previous coronary revascularization, age, gender, history of diabetes, history of β-blocker use, there was no interaction between the use of ivabradine and outcomes.
Table 1 summarizes the most significant randomized trials of ivabradine in stable CAD.
Discussion
Ivabradine represents a novel agent that decreases heart rate by inhibition of the If current channels, a mechanism different from that of β-blockers and calcium channel blockers. Ivabradine is well known to improve cardiovascular outcomes in patients with systolic heart failure (including ischemic cardiomyopathy), as demonstrated in the BEAUTIFUL and SHIFT trials [14•]. However, the addition of ivabradine to standard background anti-anginal therapy failed to demonstrate improvement in cardiovascular outcomes (e.g., cardiovascular-related death and/or non-fatal myocardial infarction). This raised a question of why do we benefit from heart rate reduction in heart failure patients but not in patients with stable CAD with normal left ventricular systolic function.
Ivabradine in lower doses contrary to the dose used in signify is currently considered a second-line, class IIa anti-anginal agent according to the latest 2013 European Society of Cardiology guidelines for the management of stable CAD [15], due to its efficacy for relieving symptoms related to angina, reduction in the number of angina attacks per week or month and improvement in quality of life. However, after getting negative outcome in the SIGNIFY trial among patients with stable CAD, the role of ivabradine is controversial in this patient population.
One of the potential explanations of this difference in outcomes between the stable CAD population in the SIGNIFY trial and the heart failure population in the SHIFT trial may reflect the fact that an elevated heart rate is due to the different pathophysiological mechanisms involved in these two disease entities. Perhaps the presence of neuro-hormonal activation in heart failure patients favored its use in this population. Also important to highlight is the fact that the dose used in the SIGNIFY trial was higher than the dose used in the SHIFT trial [16], 60 % of the patients in the shift trial received inadequate β-blockade and that most of the benefit associated with ivabradine was found in people who could not take or who were taking lower doses of β-blockers [16]. This calls into question for an optimal dose of ivabradine in combination with a β-blocker among stable CAD patients.
It is important to note the lower cardiovascular risk among the patient population at baseline in the SIGNIFY trial versus the SHIFT trial (1.4 vs 8.0 %). As it is difficult to show net mortality benefit in any trial among patients with low cardiovascular risk at baseline, perhaps one of the potential explanation for the negative outcome in the SIGNIFY trial [17].
A recent meta-analysis reported a significant increase (15 %) risk of atrial fibrillation associated with ivabradine, a finding similar to what was reported in signify trial. Although atrial fibrillation is common arrhythmia in patients with CAD and ischemic cardiomyopathy, this pro-arrythmogenic effect of ivabradine should be kept in mind while prescribing this agent [18].
Physicians should exercise caution among patients with more severe forms of stable angina and to consider adjusting β-blocker doses to effective levels before initiating ivabradine.
In the SIGNIFY trial, improved quality of life was seen with ivabradine at a heart rate of 60 to 70 beats/min; therefore, further studies are warranted in order to explore this particular subgroup of patients along with more conservative doses (e.g., 5 to 7.5 mg) that may derive clinical benefit and reduction in cardiovascular events from this agent.
After learning from the SIGNIFY and SHIFT trials, we have concluded that lowering heart rate improves outcomes in heart failure patients, positively affecting left ventricular remodeling. However, heart rate lowering in stable CAD patients without heart failure does not improve mortality outcomes as the mechanism of heart rate elevation could be different in this subgroup of patients. These findings deserve further investigations, studies, and close analyses to highlight the potential cardiovascular benefits of ivabradine.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Canty Jr JM. Coronary pressure-function and steady-state pressure-flow relations during autoregulation in the un-anesthetized dog. Circ Res. 1988;63(4):821–36.
Boudoulas H, Rittgers SE, Lewis RP, et al. Changes in diastolic time with various pharmacologic agents: implication for myocardial perfusion. Circulation. 1979;60(1):164–9.
Axsom K, Bangalore S. Heart rate in coronary artery disease: should we lower it? Curr Treat Options Cardiovasc Med. 2013;15(1):118–28.
Bohm M, Reil JC, Deedwania P, et al. Resting heart rate: risk indicator and emerging risk factor in cardiovascular disease. Am J Med. 2015;128(3):219–28.
Levine HJ. Rest heart rate and life expectancy. J Am Coll Cardiol. 1997;30(4):1104–6.
Kannel WB, Kannel C, Paffabarger RS, et al. Heart rate and cardiovascular mortality: the Framingham study. Am Heart J. 1987;113(6):1489–94.
Sulfi S, Timmis AD. Ivabradine: the first selective sinus node If channel inhibitor in the treatment of stable angina. Int J Clin Pract. 2006;60(2):222–8.
DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106(3):434–46.
Borer JS, Fox K, Jaillon P, et al. Antianginal and anti-ischemic effects of ivabradine, an If inhibitor, in stable angina: a randomized, double blind, multicenter, placebo-controlled trial. Circulation. 2003;107(3):817–23.
Tardif JC, Ford I, Tendera M, et al. Efficacy of ivabradine, a new selective If inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26(23):2529–36.
Ruzyllo W, Tendera M, Ford I, et al. Antianginal efficacy and safety of ivabradine compared with amlodipine in patients with stable effort angina pectoris: a 4-month randomized, double-blind, multicenter, non-inferiority trial. Drugs. 2007;67(3):393–405.
Tardif JC, Ponikowski P, Kahan T. Efficacy of the If current inhibitor ivabradine in patients with chronic stable angina receiving beta blocker therapy: a 4-month, randomized, placebo-controlled trial. Eur Heart J. 2009;30(5):540–8.
Fox K, Ford I, Steg PG, et al. Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med. 2014;371(12):1091–9. This original communication reports the results of the SIGNIFY trial, in which the effects of ivabradine on cardiovascular events in patients with stable coronary artery disease were analyzed.
Fox K, Komajda M, Ford I, et al. Effect of ivabradine in patients with left ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trial. Eur Heart J. 2013;34(29):2263–70. This pooled analysis demonstrated that ivabradine may be important for the improvement of clinical cardiovascular outcomes in patients with systolic heart failure.
Montalescot G, Sechtem U, Achenbach S, et al. European Society of Cardiology (ESC) guidelines on the management of stable coronary artery disease. Eur Heart J. 2013;34(38):2949–3003.
Swedberg K, Komjada M, Bohm M, et al. Effects on outcomes of heart rate reduction by ivabradine in patients with congestive heart failure: is there an influence of beta-blocker dose?—findings from the SHIFT study. J Am Coll Cardiol. 2012;59(22):1938–45.
Ferrari R, Kim KM. The role of heart rate may differ according to pathophysiological setting: from SHIFT to SIGNIFY. Eur Heart J. 2015;36(31):2042–6.
Martin RI, Pogoryelova O, Koref MS, et al. Atrial fibrillation associated with ivabradine treatment: meta-analysis of randomized controlled trials. Heart. 2014;100(7):1506–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Mateo Porres-Aguilar, Oscar C. Muñoz, and Aamer Abbas declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Ischemic Heart Disease
Rights and permissions
About this article

Cite this article
Porres-Aguilar, M., Muñoz, O.C. & Abbas, A. Current Role of Ivabradine in Stable Coronary Artery Disease Without Heart Failure. Curr Cardiol Rep 18, 13 (2016). https://doi.org/10.1007/s11886-015-0689-2
Published:
DOI: https://doi.org/10.1007/s11886-015-0689-2