
Abstract
The extent of neurologic deficit in spinal cord injury (SCI) is dependent on the primary injury, biologic responses to injury, including inflammation, edema, and scar formation, and neural restructuring. During the recovery phase of SCI, which follows a period of spinal shock lasting weeks to up to 2 years, uninhibited spinal reflexes result in detrusor overactivity with dyssynergia of the urethral sphincter, associated with progressive lower urinary tract dysfunction and potentially upper tract deterioration. Minimization of secondary injury following acute SCI and optimization of nerve regeneration may maximize functional recovery and limit end organ dysfunction, including of the lower urinary tract. Early administration of neuromodulation via electrical or electromagnetic stimulation has been shown to limit secondary injury and potentially restore function. Low-frequency electrical stimulation accelerates axonal growth in the peripheral nervous system and may have a similar benefit in the central nervous system. Limited evidence suggests that sacral neuromodulation has the potential to limit or even prevent maladaptive neural restructuring of the lower urinary tract when administered during the spinal shock phase, when the bladder is areflexic. Herein, we review the pathophysiology of voiding dysfunction following acute injury, existing evidence for the benefit of early SNM in SCI, and possible mechanisms of action, including neural regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of traumatic spinal cord injury (SCI) in the USA is 54 cases per one million persons, with an estimated 16,965 new cases in 2012 [1]. Inpatient mortality rates of 7.5 % underscore the devastating nature of SCI and associated injuries but also suggest that most patients will survive to suffer long-term consequences of their neurologic injury. Permanent sequelae of SCI may include respiratory depression, cardiovascular impairment, sensory loss, muscle spasticity, chronic pain, and bowel and bladder dysfunction [2]. However, not all consequences of SCI are destined to occur or worsen. Outcomes in SCI may be improved by limiting the pathophysiologic changes that occur following the primary injury and by augmenting the nervous system’s potentially beneficial adaptive responses [3•]. Early application of sacral neuromodulation (SNM), a procedure approved in the late 1990s by the US Food and Drug Administration (FDA) for the treatment of medication-refractory non-neurogenic overactive bladder and non-obstructive urinary retention [4], may be beneficial in limiting the progressive bladder dysfunction frequently observed following SCI [5•].
Herein, we aim to evaluate the role of early SNM in SCI. We will review the pathophysiology and epidemiology of voiding dysfunction following acute injury, existing evidence for the benefit of early SNM in SCI, and possible mechanisms of action, including neural regeneration.
Pathophysiology of Voiding Dysfunction in SCI
The spinal cord is composed of gray matter, consisting of interneurons, cell bodies and dendrites, afferent neuronal fibers, and glial cells and white matter, which consists of groups of myelinated axons [3•]. Myelinated axons travel longitudinally within the spinal cord and are responsible for transmission of signals from the periphery to the brain (i.e., ascending tracts) or from the brain to the periphery (i.e., descending tracts). Storage of urine is mediated via signaling from the pontine storage center to the pudendal and hypogastric nerves [6]. Stimulation of the pudendal nerve results in acetylcholine-mediated contraction of the external urethral sphincter (EUS), and stimulation of the post-ganglionic sympathetic hypogastric nerve results in norepinephrine-mediated inhibition of detrusor contraction and stimulation of bladder outlet contraction. As the bladder fills, increased intensity of afferent signaling from myelinated Aδ and unmyelinated C axons in the bladder stimulates the pontine micturition center (PMC) and periaqueductal gray (PAG). The PMC and PAG integrate these signals and upon release of inhibition from higher centers permit reflex voiding via efferent signaling to the sacral parasympathetic nucleus [7]. Storage-related sympathetic and pudendal nerve activity (i.e., the “guarding reflex”) is inhibited, resulting in relaxation of the bladder outlet and EUS. Parasympathetic stimulation of detrusor contraction via the pelvic nerve results in bladder emptying [6, 8, 9].
SCI disrupts signaling between the PMC and PAG and the lower urinary tract (LUT). As a result, greater than 98 % of patients with suprasacral lesions develop lower urinary tract dysfunction (LUTD) perceptible on urodynamics [10]. Detrusor overactivity (DO) is observed in 65, 78, and 49 % of patients with cervical, thoracic, and lumbar lesions, respectively [10], and greater than 40 % of patients with suprasacral SCI have poorly compliant bladders and elevated detrusor leak point pressure [11].
The extent of neurologic deficit, including LUTD, following SCI is related to the primary injury, or the direct mechanical damage associated with the initial insult, the secondary injury from the biological response to the primary injury, and a chronic phase that can result in progressive deterioration of higher centers as well as peripheral signaling [3•, 12]. Secondary injury results from hemorrhage and vasospasm leading to ischemia, free radical formation and increased inflammation, cell death resulting from apoptosis, disruption of the ionic balance, and glutamate excitotoxicity [3•]. Oligodendrocyte death and demyelination continues for up to several weeks following injury [13, 14]. Glial scar formation in the chronic phase, which can last years beyond the primary injury, inhibits axonal regeneration and remyelination, impairing functional recovery [3•, 13, 15]. Plasticity, which refers to the capacity of the central nervous system to modify its function and structure, is also critically important in the chronic phase. In the long term, plasticity is crucial to both functional recovery and progression of undesired clinical symptoms and is especially relevant to the pathophysiology of progressive deterioration of LUT function [16].
The insults associated with primary and secondary injury in suprasacral SCI lead to a period of spinal shock typically lasting 6 to 12 weeks and potentially up to 2 years in cases of complete SCI [10, 17]. Absence of reflexes below the level of the lesion, including the micturition reflex, and detrusor areflexia with maintained bladder neck tone is characteristic of spinal shock [18]. Over time, spinal reflexes return, uninhibited by efferent signaling from higher centers. The frequent result is a spastic, non-relaxing EUS combined with DO, also known as detrusor-sphincter dyssynergia (DSD). Over time, DSD is associated with elevated detrusor pressures, incontinence, urinary retention, and in many cases renal insufficiency due to progressive upper urinary tract deterioration [7, 19].
If the extent of neurologic deficit in SCI is to be minimized, early intervention, during the period when secondary injury, glial scar formation, and structural remodeling are occurring, is paramount. One such example is the benefit of aggressive early physical rehabilitation, which has been demonstrated to markedly improve functional recovery in patients with incomplete SCI [20]. There are many theories regarding mechanisms that may promote beneficial remodeling of the nervous system after SCI. These include inhibition of glial scar formation [15], reduction in the initial inflammatory response and resultant secondary injury, and cellular and molecular therapy and bioengineering to facilitate axonal growth and remyelination [3•, 21, 22]. Specific to LUTD, SNM can effectively treat symptoms of neurogenic bladder secondary to chronic SCI in select patients [23] but when administered early may alter progression of LUTD or limit its development via several of these mechanisms.
Sacral Neuromodulation in Spinal Cord Injury
Tanagho and Schmidt are credited with developing SNM for clinical application in the 1980s, demonstrating improvement in continence and emptying in patients with neuropathic voiding dysfunction [24, 25]. An implantable pulse generator designed for continuous or cycled stimulation of the third sacral nerve at a frequency of 10–14 Hz was ultimately approved by the FDA in 1997 for the treatment of urge urinary incontinence and in 1999 for urinary frequency and non-obstructive urinary retention [4]. Despite its efficacy and widespread use, the mechanism of action of SNM is not known. It is postulated to act on both the peripheral and central nervous systems [26], with its effects possibly enhanced over time by neural plasticity [27].
It has traditionally been believed that intact spinal pathways are required in order for SNM to be effective, and in general, SNM has not been found to be as effective in SCI [28]. Approximately 40 % of patients with neurogenic LUTD secondary to chronic (>6 month) incomplete SCI undergoing unilateral or bilateral SNM proceeded to permanent implant based on >50 % improvement in symptoms [29, 30]. When initially effective, however, results appear to be durable, with sustained benefit in LUT symptoms observed at a median follow-up of 61 months after SNM [23]. Furthermore, subsequent studies have reported permanent implant rates as high as 70 % [31, 32], as well as the potential for benefit in patients with neurogenic non-obstructive retention secondary to incomplete SCI [32, 33]. In contrast, multiple reports [28, 34•, 35] failed to demonstrate efficacy of SNM in patients with chronic complete SCI.
Each of these studies, however, represented efforts to control refractory symptoms of neurogenic bladder in the chronic SCI patient. A 1998 study by Shaker et al. suggested the potential for SNM to prevent neurogenic LUTD altogether when administered in the acute phase of SCI [36]. The authors placed sacral nerve stimulators in 11 dogs following complete spinal cord transection at the T10 level and were able to demonstrate selective blockade of the external urethral sphincter and stimulation of detrusor contraction. With chronic stimulation, the authors were able to maintain complete emptying in over half of the dogs, although these changes were not maintained in the absence of stimulation [37].
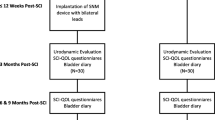
In a prospective study representing the first to evaluate percutaneous SNM in the acute SCI phase in humans, Sievert et al. implanted tined leads bilaterally into the third sacral foramina of ten patients with complete SCI above T12 [5•]. Six patients declined to participate but were followed as controls and received usual care with antimuscarinics. All patients were confirmed to have detrusor acontractility prior to lead implantation. Leads were placed at a mean of 2.9 months following the initial injury, and each was connected to a separate implantable pulse generator. At a mean follow-up of 26.2 months, patients who underwent SNM had higher average catheterization volumes (582 vs. 208 mL), had experienced fewer UTIs (0.5 vs. 3.8), and appeared to have improved compliance and continence relative to controls, although no direct statistical head to head comparisons were performed. Five patients who underwent SNM and experienced malfunction of the device due to lead failure experienced DO with incontinence that improved with lead revision.
As a small, non-randomized study, the study by Sievert and colleagues is primarily hypothesis generating. It is nevertheless intriguing, as prevention of DO, DSD, and elevated detrusor pressures would be a remarkable development in the urologic care of patients with SCI. Further study may be guided by exploration of the possible mechanisms by which early SNM may optimize recovery and prevent LUT deterioration rather than simply treat symptoms in patients whose neurourologic fate may already be determined.
Mechanism of Action
Functional recovery after SCI is related to minimizing secondary injury and maladaptive chronic phase neural restructuring as well as axonal growth and remyelination [3•, 22, 38]. In chronic SCI, SNM may prevent maladaptive restructuring of neural circuitry through alteration of afferent signaling via sympathetic pathways, which are relatively spared in SCI below T7/T8 [16]. Intact ascending pathways may also play a role in incomplete SCI [5•, 39]. Even in the case of complete SCI, in which there is complete absence of intact descending parasympathetic signaling, it is possible that sympathetic signaling could inhibit parasympathetic activity in the pelvic ganglia [40]. Early SNM may also prevent upregulation of “silent” C-fiber-mediated reflex pathways whose unmasking contributes significantly to DO in chronic SCI [38, 41].
Neuromodulation may also promote axonal growth and myelination, both of which are correlated with functional recovery [13, 22]. Epidural stimulation via surgically placed dural leads and extensive locomotor training has been associated with restoration of neural circuitry permitting proprioception, balance, and stepping in humans [42, 43] and voiding in rats [44]. Non-invasive neuromodulation via extremely low frequency electromagnetic field stimulation has been suggested to promote axonal growth by reducing inhibition of the corticospinal tract, permitting formation of new functional intraspinal circuits [45]. Electromagnetic field stimulation may also optimize conditions for axonal recovery via optimization of neurotransmitter levels, promoting angiogenesis, and increasing neurotrophic factors [46–49]. Similarly, transcutaneous electrical stimulation of the brain and spinal cord, another non-invasive form of neuromodulation, has been suggested to promote functional recovery and neuroplasticity [50].
It is plausible that electrical stimulation via SNM might also promote axonal growth; however, the evidence to suggest this is primarily limited to the peripheral nervous system. SNM has been demonstrated to modify nerve growth factor (NGF) levels in the urine in patients with non-neurogenic overactive bladder deriving symptomatic benefit, suggesting its potential to modify other nerve growth factors peripherally and centrally [51]. Increased levels of brain-derived neurotrophic factor (BDNF) have been demonstrated with electrical stimulation of peripheral nerves in multiple settings [52]. It is possible that SNM augments the activity and/or increases levels of BDNF and other neurotrophic factor which promotes axonal growth in the central nervous system after SCI [53••]. Low frequency (20 Hz) electrical stimulation is well-studied in the peripheral nervous system, where it has been demonstrated to accelerate axonal growth [54, 55]. Twenty hertz peripheral electrical stimulation may also speed sensory nerve regeneration in the central nervous system [56].
Administration of electrical stimulation early after nerve injury is important to maximize benefit in the peripheral nervous system because the inherent ability of nerves to regenerate decreases with time [52, 57]. Therefore, it is likely that early SNM would be required to promote nerve regeneration in the central nervous system as well, provided that response to electrical stimulation in the central nervous system parallels the response in the peripheral nervous system. However, inherent differences between axonal support cells, response to neurotrophic factors, and signaling in the central and peripheral nervous systems may limit this assumption [55].
Conclusions
A single study demonstrates a clinical benefit for early SNM following SCI. Though encouraging, validation in larger studies is required, and exploration of potential mechanisms is warranted. Early SNM may promote functional recovery of voiding function following SCI by reducing the extent of secondary injury and maladaptive neural restructuring. Low frequency electrical stimulation stimulates peripheral nerve re-growth, and invasive and non-invasive electrical and electromagnetic stimulation may promote axonal regeneration in the central nervous system following SCI via upregulation of neurotrophic factors and increased angiogenesis. It is therefore plausible that SNM promotes axonal growth in the central nervous system after SCI; however, in the absence of further evidence, this theory is highly speculative. Continued study of the mechanisms of action of SNM may provide further insight and guide future clinical studies of early SNM in acute phase SCI.
BDNF, brain-derived neurotrophic factor; DO, detrusor overactivity; DSD, detrusor-sphincter dyssynergia; EUS, external urethral sphincter; LUT, lower urinary tract; LUTD, lower urinary tract dysfunction; NGF, nerve growth factor; PAG, periaqueductal gray; PMC, pontine micturition center; SCI, spinal cord injury; SNM, sacral neuromodulation
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jain NB, Ayers GD, Peterson EN, Harris MB, Morse L, O’Connor KC, et al. Traumatic spinal cord injury in the United States, 1993–2012. JAMA. 2015;313:2236–43. doi:10.1001/jama.2015.6250.
Sezer N, Akkuş S, Uğurlu FG. Chronic complications of spinal cord injury. World J Orthop. 2015;6:24–33. doi:10.5312/wjo.v6.i1.24.
Silva NA, Sousa N, Reis RL, Salgado AJ. From basics to clinical: a comprehensive review on spinal cord injury. Prog Neurobiol. 2014;114:25–57. doi:10.1016/j.pneurobio.2013.11.002. The authors present an excellent overview of the pathophysiology of SCI and evolving therapies in the acute and chronic phases of injury.
Liberman D, Ehlert MJ, Siegel SW. Sacral neuromodulation in urological practice. Urology n.d. doi:10.1016/j.urology.2016.06.004.
Sievert K-D, Amend B, Gakis G, Toomey P, Badke A, Kaps HP, et al. Early sacral neuromodulation prevents urinary incontinence after complete spinal cord injury. Ann Neurol. 2010;67:74–84. doi:10.1002/ana.21814. This represents the only study in humans to demonstrate prevention of neurogenic detrusor overactivity by early administration of SNM. The authors prospectively followed 10 patients with complete SCI following bilateral S3 lead placement a mean of 2.9 months after the initial injury and 6 who declined therapy.
Chai TC, Birder LA. Physiology and pharmacology of the bladder and urethra. In: Wein AJ, Kavoussi LR, Partin AW, Peters CA, editors. Campbell-Walsh Urol. 11th Ed., vol. 3. Philadelphia: Elsevier; 2016. p. 1631–84.
Fowler CJ, Griffiths D, de Groat WC. The neural control of micturition. Nat Rev Neurosci. 2008;9:453–66. doi:10.1038/nrn2401.
Griffiths DJ, Fowler CJ. The micturition switch and its forebrain influences. Acta Physiol (Oxford, England). 2013;207:93–109. doi:10.1111/apha.12019.
de Groat WC, Wickens C. Organization of the neural switching circuitry underlying reflex micturition. Acta Physiol (Oxford, England). 2013;207:66–84. doi:10.1111/apha.12014.
Jeong SJ, Cho SY, Oh S-J. Spinal cord/brain injury and the neurogenic bladder. Urol Clin N Am. 2010;37:537–46. doi:10.1016/j.ucl.2010.06.005.
Weld KJ, Dmochowski RR. Association of level of injury and bladder behavior in patients with post-traumatic spinal cord injury. Urology. 2000;55:490–4. doi:10.1016/S0090-4295(99)00553-1.
Cramer SC, Lastra L, Lacourse MG, Cohen MJ. Brain motor system function after chronic, complete spinal cord injury. Brain J Neurol. 2005;128:2941–50. doi:10.1093/brain/awh648.
Plemel JR, Keough MB, Duncan GJ, Sparling JS, Yong VW, Stys PK, et al. Remyelination after spinal cord injury: is it a target for repair? Prog Neurobiol. 2014;117:54–72. doi:10.1016/j.pneurobio.2014.02.006.
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–6.
Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J. Functional regeneration beyond the glial scar. Exp Neurol. 2014;253:197–207. doi:10.1016/j.expneurol.2013.12.024.
Wein AJ, Dmochowski RR. Neuromuscular dysfunction of the lower urinary tract. In: Wein AJ, Kavoussi LR, Partin AW, Peters CA, editors. Campbell-Walsh Urol. 11th Ed., vol. 3. Philadelphia: Elsevier; 2016. p. 1761–95.
Samson G, Cardenas DD. Neurogenic bladder in spinal cord injury. Phys Med Rehabil Clin N Am. 2007;18:255–74. doi:10.1016/j.pmr.2007.03.005.
Rossier AB, Fam BA, Dibenedetto M, Sarkarati M. Urodynamics in spinal shock patients. J Urol. 1979;122:783–7.
Patki P, Woodhouse J, Hamid R, Shah J, Craggs M. Lower urinary tract dysfunction in ambulatory patients with incomplete spinal cord injury. J Urol. 2006;175:1784–7. doi:10.1016/S0022-5347(05)00979-1.
Wernig A, Müller S, Nanassy A, Cagol E. Laufband therapy based on “rules of spinal locomotion” is effective in spinal cord injured persons. Eur J Neurosci. 1995;7:823–9. doi:10.1111/j.1460-9568.1995.tb00686.x.
Ahuja CS, Fehlings M. Concise review: bridging the gap: novel neuroregenerative and neuroprotective strategies in spinal cord injury. Stem Cells Transl Med. 2016;5:914–24. doi:10.5966/sctm.2015-0381.
Wu B, Ren X. Promoting axonal myelination for improving neurological recovery in spinal cord injury. J Neurotrauma. 2009;26:1847–56. doi:10.1089/neu.2008.0551.
Lombardi G, Del Popolo G. Clinical outcome of sacral neuromodulation in incomplete spinal cord injured patients suffering from neurogenic lower urinary tract symptoms. Spinal Cord. 2009;47:486–91. doi:10.1038/sc.2008.172.
Tanagho EA, Schmidt RA. Bladder pacemaker: scientific basis and clinical future. Urology. 1982;20:614–9.
Tanagho EA, Schmidt RA, Orvis BR. Neural stimulation for control of voiding dysfunction: a preliminary report in 22 patients with serious neuropathic voiding disorders. J Urol. 1989;142:340–5.
Amend B, Matzel KE, Abrams P, de Groat WC, Sievert K-D. How does neuromodulation work. Neurourol Urodyn. 2011;30:762–5. doi:10.1002/nau.21096.
van der Pal F, Heesakkers JPFA, Bemelmans BLH. Current opinion on the working mechanisms of neuromodulation in the treatment of lower urinary tract dysfunction. Curr Opin Urol. 2006;16:261–7. doi:10.1097/01.mou.0000232047.87803.1e.
Hohenfellner M, Humke J, Hampel C, Dahms S, Matzel K, Roth S, et al. Chronic sacral neuromodulation for treatment of neurogenic bladder dysfunction: long-term results with unilateral implants. Urology. 2001;58:887–92.
Lombardi G, Nelli F, Mencarini M, Del Popolo G. Clinical concomitant benefits on pelvic floor dysfunctions after sacral neuromodulation in patients with incomplete spinal cord injury. Spinal Cord. 2011;49:629–36. doi:10.1038/sc.2010.176.
Lombardi G, Musco S, Celso M, Ierardi A, Nelli F, Del Corso F, et al. Intravesical electrostimulation versus sacral neuromodulation for incomplete spinal cord patients suffering from neurogenic non-obstructive urinary retention. Spinal Cord. 2013;51:571–8. doi:10.1038/sc.2013.37.
Chen G, Liao L. Sacral neuromodulation for neurogenic bladder and bowel dysfunction with multiple symptoms secondary to spinal cord disease. Spinal Cord. 2014. doi:10.1038/sc.2014.157.
Wöllner J, Krebs J, Pannek J. Sacral neuromodulation in patients with neurogenic lower urinary tract dysfunction. Spinal Cord. 2016;54:137–40. doi:10.1038/sc.2015.124.
Lombardi G, Musco S, Celso M, Del Corso F, Del Popolo G. Sacral neuromodulation for neurogenic non-obstructive urinary retention in incomplete spinal cord patients: a ten-year follow-up single-centre experience. Spinal Cord. 2014;52:241–5. doi:10.1038/sc.2013.155.
Gaunt RA, Prochazka A. Control of urinary bladder function with devices: successes and failures. Prog Brain Res. 2006;152:163–94. doi:10.1016/S0079-6123(05)52011-9. The authors present compelling animal data that early electrical stimulation of the sacral root may have the capacity to maintain voiding after SCI and prevent DSD.
Schurch B, Reilly I, Reitz A, Curt A. Electrophysiological recordings during the peripheral nerve evaluation (PNE) test in complete spinal cord injury patients. World J Urol. 2003;20:319–22. doi:10.1007/s00345-002-0299-7.
Shaker HS, Tu LM, Robin S, Arabi K, Hassouna M, Sawan M, et al. Reduction of bladder outlet resistance by selective sacral root stimulation using high-frequency blockade in dogs: an acute study. J Urol. 1998;160:901–7.
Abdel-Gawad M, Boyer S, Sawan M, Elhilali MM. Reduction of bladder outlet resistance by selective stimulation of the ventral sacral root using high frequency blockade: a chronic study in spinal cord transected dogs. J Urol. 2001;166:728–33. doi:10.1016/S0022-5347(05)66051-X.
de Groat WC, Yoshimura N. Plasticity in reflex pathways to the lower urinary tract following spinal cord injury. Exp Neurol. 2012;235:123–32. doi:10.1016/j.expneurol.2011.05.003.
Craggs M, McFarlane J. Neuromodulation of the lower urinary tract. Exp Physiol. 1999;84:149–60.
de Groat WC, Booth AM. Inhibition and facilitation in parasympathetic ganglia of the urinary bladder. Fed Proc. 1980;39:2990–6.
de Groat WC, Yoshimura N. Mechanisms underlying the recovery of lower urinary tract function following spinal cord injury. Prog Brain Res. 2006;152:59–84. doi:10.1016/S0079-6123(05)52005-3.
Gerasimenko Y, Roy RR, Edgerton VR. Epidural stimulation: comparison of the spinal circuits that generate and control locomotion in rats, cats and humans. Exp Neurol. 2008;209:417–25. doi:10.1016/j.expneurol.2007.07.015.
Harkema S, Gerasimenko Y, Hodes J, Burdick J, Angeli C, Chen Y, et al. Effect of epidural stimulation of the lumbosacral spinal cord on voluntary movement, standing, and assisted stepping after motor complete paraplegia: a case study. Lancet. 2011;377:1938–47. doi:10.1016/S0140-6736(11)60547-3.
Gad PN, Roy RR, Zhong H, Lu DC, Gerasimenko YP, Edgerton VR. Initiation of bladder voiding with epidural stimulation in paralyzed, step trained rats. PLoS ONE. 2014;9:e108184. doi:10.1371/journal.pone.0108184.
Kumar S, Dey S, Jain S. Extremely low-frequency electromagnetic fields: a possible non-invasive therapeutic tool for spinal cord injury rehabilitation. Electromagn Biol Med. 2016:1–14. doi:10.1080/15368378.2016.1194290.
Kumar S, Jain S, Velpandian T, Petrovich Gerasimenko Y, Avelev V, Behari J, et al. Exposure to extremely low-frequency magnetic field restores spinal cord injury-induced tonic pain and its related neurotransmitter concentration in the brain. Electromagn Biol Med. 2013;32:471–83. doi:10.3109/15368378.2012.743907.
Delle Monache S, Alessandro R, Iorio R, Gualtieri G, Colonna R. Extremely low frequency electromagnetic fields (ELF-EMFs) induce in vitro angiogenesis process in human endothelial cells. Bioelectromagnetics. 2008;29:640–8. doi:10.1002/bem.20430.
Delle Monache S, Angelucci A, Sanità P, Iorio R, Bennato F, Mancini F, et al. Inhibition of angiogenesis mediated by extremely low-frequency magnetic fields (ELF-MFs). PLoS ONE. 2013;8:e79309. doi:10.1371/journal.pone.0079309.
Mert T, Gunay I, Gocmen C, Kaya M, Polat S. Regenerative effects of pulsed magnetic field on injured peripheral nerves. Altern Ther Health Med. 2006;12:42–9.
Nardone R, Höller Y, Taylor A, Thomschewski A, Orioli A, Frey V, et al. Noninvasive spinal cord stimulation: technical aspects and therapeutic applications. Neuromodulation Technol Neural Interface. 2015;18:580–91. doi:10.1111/ner.12332.
Shalom DF, Pillalamarri N, Xue X, Kohn N, Lind LR, Winkler HA, et al. Sacral nerve stimulation reduces elevated urinary nerve growth factor levels in women with symptomatic detrusor overactivity. Am J Obstet Gynecol 2014;211:561.e1–561.e5. doi:10.1016/j.ajog.2014.07.007.
Gordon T. Electrical stimulation to enhance axon regeneration after peripheral nerve injuries in animal models and humans. Neurother J Am Soc Exp Neurother. 2016;13:295–310. doi:10.1007/s13311-015-0415-1.
Frias B, Santos J, Morgado M, Sousa MM, Gray SMY, McCloskey KD, et al. The role of brain-derived neurotrophic factor (BDNF) in the development of neurogenic detrusor overactivity (NDO). J Neurosci Off J Soc Neurosci. 2015;35:2146–60. doi:10.1523/JNEUROSCI.0373-14.2015. This review from one of the world’s experts in nerve regeneration describes the differences between mechanisms of regrowth in the peripheral and central nervous systems. This review highlights some of the most insightful and important studies in the field to date.
Al-Majed AA, Neumann CM, Brushart TM, Gordon T. Brief electrical stimulation promotes the speed and accuracy of motor axonal regeneration. J Neurosci Off J Soc Neurosci. 2000;20:2602–8.
Gordon T. Nerve regeneration in the peripheral and central nervous systems. J Physiol. 2016;594:3517–20. doi:10.1113/JP270898.
Gordon T, Udina E, Verge VMK, de Chaves EIP. Brief electrical stimulation accelerates axon regeneration in the peripheral nervous system and promotes sensory axon regeneration in the central nervous system. Mot Control. 2009;13:412–41.
Lin Y-C, Kao C-H, Chen C-C, Ke C-J, Yao C-H, Chen Y-S. Time-course effect of electrical stimulation on nerve regeneration of diabetic rats. PLoS ONE. 2015;10:e0116711. doi:10.1371/journal.pone.0116711.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs Cohn, Kowalik, Milam, and Reynolds declare no conflicts of interest.
Dr. Kaufman reports other from Medtronic, during the conduct of the study.
Dr. Dmochowski reports other from Allergan, other from Medtronic, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain studies with human or animal subjects performed by the author.
Additional information
This article is part of the Topical Collection on Neurogenic Bladder
Rights and permissions
About this article

Cite this article
Cohn, J.A., Kaufman, M.R., Dmochowski, R.R. et al. Early Sacral Neuromodulation in Spinal Cord Injury—Can It Regenerate Nerves?. Curr Bladder Dysfunct Rep 11, 350–355 (2016). https://doi.org/10.1007/s11884-016-0382-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11884-016-0382-3