
Abstract
Purpose of Review
Elevated low-density lipoprotein cholesterol (LDL-C) and triglyceride-rich lipoproteins (TRLs) or remnants are important risk factors for the development of atherosclerotic cardiovascular disease (ASCVD). The ongoing challenge of not being able to achieve recommended LDL-C targets despite maximally tolerated lipid-lowering therapy (LLT) has led to the development of novel therapeutic agents including angiopoietin-like 3 (ANGPTL3) inhibitors.
Recent Findings
ANGPTL3 is a glycoprotein produced by the liver that inhibits lipoprotein lipase and endothelial lipase. Data from genetic and clinical studies have shown that a lower ANGPTL3 level is associated with lower plasma LDL-C, triglyceride (TG), and other lipoproteins. Pharmacological inactivation of ANGPTL3 with the monoclonal antibody, evinacumab, results in a 50% reduction in LDL-C, even in patients with homozygous familial hypercholesterolemia (HoFH). The safe and effective targeted delivery of nucleic acid–based therapies will shape the future of the lipid arena.
Summary
ANGPTL3 is a novel target in lipoprotein metabolism, targeting not only LDL-C via an LDL-receptor (LDLR) independent mechanism but also TRLs and carries a significant promise for further ASCVD risk reduction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerotic cardiovascular disease (ASCVD) is a leading cause of morbidity and mortality worldwide with dyslipidemia being an important risk factor. Epidemiological and observational studies have shown a clear association between elevated atherogenic plasma lipoproteins, particularly low-density lipoprotein cholesterol (LDL-C), triglyceride-rich lipoproteins (TRLs), lipoprotein(a) (Lp(a)), and ASCVD [1]. Statins are the mainstay of therapy for lowering LDL-C, and evidence from large randomized controlled trials has shown that for every 40-mg/dL (1 mmol/L) reduction in LDL-C, about a one-fifth reduction in stroke, major coronary events, and revascularization is seen with no lower limit and no safety issues [2]. Hence, it is clear with regards to LDL-C that “lower is better.” Most drug therapies in current use, such as statins, ezetimibe, and bempedoic acid, are LDL-receptor (LDLR) dependent and rely on upregulation of the LDLR for their action. Individuals with familial hypercholesterolemia (FH), with genetic mutations in the LDLR, especially those with homozygous FH (HoFH) who have minimal on no residual LDLR function, will benefit from therapeutic interventions acting via a LDLR-independent mechanism. In addition, despite advances made with LDL-C targeted lipid-lowering therapy (LLT), residual ASCVD risk remains high and elevated TG levels, specifically TRLs or remnants, may contribute to this risk [3, 4••, 5, 6•]. Further therapeutic modulation of LDL-C as well as triglyceride (TG) is thus the new focus of LLT, with angiopoietin-like 3 (ANGPTL3) inhibition emerging as an important therapeutic strategy in lowering LDL-C as well as TG via a LDLR-independent pathway.
Role of ANGPTL3 in Lipid Metabolism
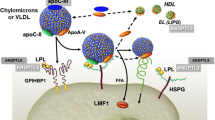
The TRLs arise from either endogenous or exogenous sources of TG. Chylomicrons contain apolipoprotein (apo) B48 and are synthesized in the intestine. Very low-density lipoprotein (VLDL), containing apoB100, is produced in the liver and comprises TG made from free fatty acids and glycerol [6•, 7•]. Further apolipoproteins (apoC-1, apoC-II, apoC-III, and apoE) are added to the surface of VLDL before being secreted by the liver into the circulation [7•]. Chylomicrons and VLDL are then hydrolyzed by lipoprotein lipase (LPL) into smaller particles of VLDL, intermediate-density lipoprotein (IDL), and LDL [6•]. LPL is produced by adipocytes and myocytes and is expressed on cell surfaces by heparin sulfate proteoglycans (HSPG) following which glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) transports LPL to the capillary endothelium where it exerts effects on circulating TRL [8, 9]. LPL, and thus TG metabolism, is highly regulated by apoC-II, apoC-III, apoA-V, ANGPTL3, ANGPTL4, and ANGPTL8 [10,11,12].
Biology of ANGPTL3
The angiopoietin-like (ANGPTL) proteins, comprising eight members, are structurally homologous to angiopoietins but do not act on the endothelial tyrosine kinase receptors, TIE1 and TIE2, to induce angiogenesis [12]. Like the angiopoietins, they contain a signal peptide of 16 amino acids, an N-terminal coiled coil domain, a linker region, and a fibrinogen-like domain at the C-terminal [13]. The N-terminal coiled coil domain is responsible for the inhibition of LPL and endothelial lipase (EL), while the C-terminal domain has a role in angiogenesis [12, 14•].
In 1999, ANGPTL3, a 70-kDa glycoprotein comprising 460 amino acids, was discovered and found to be expressed in the liver of both mice and humans [15]. Its potential role in lipid metabolism was considered following observations that homozygous mutations in ANGPTL3 within a substrain of KK mice (termed KK/San), a murine model of moderate obesity, hyperglycemia, and hyperinsulinemia, were found to have hypolipidemia [16]. Administration of the ANGPTL3 protein to KK/San mice subsequently led to a rise in serum lipid fractions [16].
Cleavage of ANGPTL3 at the linker region occurs both intracellularly by the proprotein convertase furin (also known as PCSK3) and extracellularly by paired amino acid–converting enzyme (PACE4 or PCSK6) [13, 17]. O-Glycosylation by N-acetylgalactosaminyltransferase 2 (GalNAcT2) occurs next to the cleavage site of ANGPTL3 which inhibits proprotein convertase-mediated cleavage and may modulate the activation of ANGPTL3 [18]. The N-terminal domain is the portion responsible for the inhibitory activity on LPL; however, whether cleavage is entirely necessary for LPL inhibition is not entirely clear [13, 19•]. However, the truncated form of ANGPTL3 appears to be more active [13, 20].
ANGPTL3 function is enhanced by the formation of a complex with ANGPTL8 [12]. This occurs in the liver and leads to inhibition of LPL to a degree far greater than either ANGPTL3 or ANGPTL8 alone [20]. The ANGPTL3/8 complex inhibits LPL by furin-mediated cleavage, enabling unfolding and dissociation of LPL [7•, 21•]. The expression of ANGPTL3 in the liver is not significantly affected by the fasting or fed state; however, the effects of ANGPTL8 on LPL activity are more prominent after eating [12]. Mechanisms by which ANGPTL3 inhibits EL remain unclear.
The inhibitory effects of ANGPTL3 and the ANGPTL3/8 complex on LPL activity are only minimally affected by prolonged exposure to cold [22]. This is in contrast to the ANGPTL4/8 complex which has increased LPL activity in brown adipose tissue with prolonged exposure to cold, suggesting a role in thermogenesis [22].
Genetic Variants of ANGPTL3
In 1991, Fazio reported on a family with familial hypobetalipoproteinemia (FHBL), an autosomal dominant disorder characterized by markedly low serum VLDL, IDL, and LDL-C levels, that was not due to a mutation in the gene encoding for apo B [23]. Later studies revealed mutations in the ANGPTL3 gene, located on chromosome 1p31.1, to be the cause and the first loss-of-function (LOF) mutation in humans was described in a family of European descent [15, 24].
A number of genome-wide association studies (GWAS) have identified significant associations between loss of function mutations in ANGPTL3 and lower TG, HDL-C, and LDL-C. Whole-exome sequencing of 58,335 individuals identified 13 distinct LOF mutations in ANGPTL3 among 246 individuals [25]. Those who carried these mutations, all of whom were heterozygotes, had a 27% lower TG, 9% lower LDL-C, and 4% lower HDL-C as compared to non-carriers [25]. Homozygous mutations in ANGPTL3, on the other hand, are associated with a 67% reduction in LDL-C, 71% reduction in TG, and a 39% reduction in HDL-C [26]. Reduction in LDL-C due to ANGPTL3 deficiency can be explained by a reduction in hepatic apo B secretion as well as an increase in hepatic LDL-C uptake [27].
The exons of the ANGPTL3, ANGPTL4, and ANGPTL5 genes were sequenced in 3551 participants in the multi-ethnic Dallas Heart Study [28]. Overall, 1% of all participants and 4% falling within the lowest quartile of plasma TG were found to have a LOF mutation in one of these three genes [28]. A spectrum of both rare and common mutations, including nonsense, frameshift, splice-site, or missense mutations, were identified within this group and located within the N-terminal coiled-coil domain of ANGPTL3, the region which has an inhibitory effect on LPL [28, 29]. This finding was consistent among studies of homozygotes and compound heterozygotes with familial combined hypolipidemia.
The transcription factors liver X receptor-α (LXRα) and hepatocyte nuclear factor-1α (HNF-1α) promote the expression of the ANGPTL3 gene [30]. Leptin, thyroid hormone, and insulin reduce circulating ANGPTL3 by decreasing expression of ANGPTL3 in the liver [31, 32]. Statins reduced the mRNA expression of ANGPTL3 by around 25% in healthy subjects as well as circulating levels of ANGPTL3 in individuals with hypercholesterolemia, and this may be the reason even subjects with LDLR-negative HoFH respond partially to statin therapy [33, 34•].
Atherosclerosis and ANGPTL3
TRLs, which include chylomicrons, VLDL, and their remnants, are pro-atherogenic and are causally associated with endothelial inflammation, ASCVD, and an increase in all-cause mortally [5]. Genetic studies have shown that genetic variants in LPL and heterozygous and homozygous LOF mutations of its inhibitory co-factors including APOC3, ANGPTL3, and ANGPTL4 are associated with reductions in TG and ASCVD [35]. Compared to those without a mutation in ANGPTL3, heterozygous carriers of ANGPTL3 LOF mutations exhibited a 34% reduction in the odds of coronary artery disease [28]. Consistent with the association of ANGPTL3 and an increased risk for ASCVD, data from the PROMIS study showed that individuals in the lowest tertile of serum ANGPTL3 levels were significantly at a lower risk of a myocardial infarction as compared to those in the highest tertile [28]. This was further supported by the DiscovEHR study, whereby genetic and therapeutic antagonism of ANGPTL3 in humans and mice was associated with significantly lower levels of TG, HDL-C, and LDL-C, with 41% lower odds for ASCVD as compared to the general population [25].
Most circulating TG are carried by VLDL particles and their remnants, which also contain apo B, and data show that apo B–containing lipoproteins also increase ASCVD. Hence, ASCVD risk mediated by TRLs or remnants appears to be determined by the concentration of apo B rather than TG per se [35]. Data from a large Mendelian randomization analysis showed that LPL and LDL-C-lowering genetic variants are associated with a similar reduction in risk for ASCVD per unit difference in apo B and thus the clinical benefit in ASCVD reduction may be more likely related to the absolute reduction in apo B–containing lipoproteins [36]. Thus, therapeutic inhibition of ANGPTL3 will have the benefit of reducing not only LDL-C but also the apo B–containing TRLs with the potential of reducing residual ASCVD burden.
ANGPTL3 as a Therapeutic Target
Mechanism of Action of ANGPTL3 Inhibition
The reduction in TRLs and HDL-C by ANGPTL3 inhibition is attributed to an increased LPL and EL activity, respectively. Peripheral clearance of chylomicrons and VLDL particles is augmented by the increase in LPL activity resulting in a decrease in TG [37•]. The exact mechanism of LDL-C reduction by ANGPTL3 inhibition remains unclear. Current postulated mechanisms include an increased LPL activity mediated hydrolysis of VLDL, with an enhanced clearance and reduced production of LDL [38]. In the absence of functional LDLR activity, lowering of LDL-C appears to be dependent on EL [38]. Increasing EL activity via ANGPTL3 inhibition modifies the composition of VLDL, resulting in the formation of lipid-depleted remnant particles that are rapidly cleared from the circulation, with a resultant depletion of the hepatic LDL-C precursor pool, ultimately reducing LDL-C. In contrast, when LDLR function is preserved, the LDL-C-lowering effect of ANGPTL3 inactivation appears to be independent of EL and may be related to increased LDL uptake by the LDLR, other receptors, or via an unidentified non-receptor-mediated mechanism [39, 40••]. These notable effects of ANGPTL3 on lipid metabolism have set the stage for using ANGPTL3 as a molecular therapeutic target in the management of dyslipidemia and the prevention of ASCVD.
Pharmacological Inactivation of ANGPTL3
Pharmacological advances utilizing monoclonal antibodies or nucleic acid–based therapeutics allow for a more targeted lipid-lowering approach.
Monoclonal Antibody–Based Therapy—Evinacumab
Evinacumab is a fully human monoclonal antibody (mAb) directed against ANGPTL3, which, in preclinical studies in mice and monkeys, reversed ANGPTL3-induced inhibition of LPL, with subsequent reductions in plasma TG, LDL-C, and HDL-C [39]. Human studies were consistent with animal data, whereby the phase I single ascending dose study conducted in 83 healthy adults receiving different subcutaneous and intravenous (IV) doses showed a dose-dependent reduction in fasting TG and LDL-C of up to 63 and 28%, respectively, with IV administration being more effective [25]. A reduction in LDL-C of almost 50% was seen in the phase II study of nine patients with HoFH and was further confirmed in the ELIPSE HoFH phase III trial, whereby a 47% reduction in LDL-C and a decrease in TG and apo B of 55 and 41%, respectively, was seen in those receiving evinacumab versus placebo [40••, 41]. Almost 50% of patients achieved an LDL-C < 100 mg/dL (< 2.6 mmol/L) and up to 30% achieved an LDL-C < 70 mg/dL (< 1.8 mmol/L) [40••]. The reduction of LDL-C was similar in those HoFH patients with no residual LDLR function (null-null HoFH), thus supporting the LDLR-independent mechanism of ANGPTL3 inhibition. No major safety concerns were seen with evinacumab, and its long-term safety and efficacy have been confirmed in the open-label extension of the ELIPSE HoFH trial, whereby a 46% reduction in LDL-C was seen at week 48, with 50% of patients achieving a target LDL-C < 100 mg/dL (< 2.6 mmol/L) and 31.7% achieved an LDL-C < 70 mg/dL (< 1.8 mmol/L) [42•]. Evinacumab is thus an effective therapeutic modality for patients with HoFH who fail to achieve target LDL-C on conventional LLT including PCSK9-inhibitor therapy with or without lipoprotein apheresis. It has also been shown to benefit patients with severe heterozygous FH (HeFH) or refractory hypercholesterolemia, whereby a further 50% reduction in LDL-C as compared to placebo was seen with the maximum IV dose [43••]. The need for monthly IV dosing of evinacumab is a potential limiting factor when compared to the newer therapeutic options currently under development to inhibit ANGPTL3.
Genetic Inactivation of ANGPTL3
Nucleic acid inhibition of ANGPTL3 mRNA transcription includes antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs). ASO binds to ANGPTL3 mRNA within the nucleus to form a DNA–RNA hetero-duplex. In contrast to ASOs, with siRNA therapy, the conjugated nucleic acids are directed at controlling the translation of the cognate mRNA in the cytoplasm [37•]. Advances in nucleic acid technology have resulted in an improved delivery of ASO and siRNA to the liver with the development of N-acetylgalactosamine (GalNAc) conjugated technology, whereby GalNAc is as efficacious as unconjugated nucleic acid regimens but offers the benefit of utilizing a 30-fold lower dose of the drug, thus reducing systemic exposure [4••, 37•].
Antisense-Oligonucleotide Therapy (ASO)—Vupanorsen (IONIS-ANGPTL3-LRX)
Vupanorsen is a second-generation GalNAc-modified single-stranded RNA ASO targeted at hepatic ANGPTL3 mRNA, promoting degradation of ANGPTL3, with a resultant decrease in its hepatic production and secretion [4••, 37•]. Data from the phase I, randomized, double-blind, placebo controlled trial, designed to assess the safety of subcutaneous single (20, 40, or 80 mg) and multiple ascending doses (10, 20, 40, 60 mg for 6 weeks) of vupanorsen, showed a dose-dependent effect and a reduction in plasma TG of 50% and LDL-C of 33% at day 43, with the 60 mg SC weekly multi-dose regimen [37•]. A dose-dependent reduction of 22% in apo B and 57% in Lp(a) at day 43 was also seen; however, subjects enrolled into this study had low pre-treatment Lp(a) levels [37•, 44]. In the phase II, placebo-controlled, dose-ranging study, 105 participants with an elevated fasting TG level > 150 mg/dL (> 1.7 mmol/L), type 2 diabetes with a glycosylated hemoglobin (HbA1c) > 6.5 and ≤ 10%, hepatic steatosis, and a body mass index (BMI) 27–40 kg/m2 were treated for 6 months with either 40 or 80 mg subcutaneously every 4 weeks, or 20 mg vupanorsen weekly [4••]. There was a significant reduction in TG of 53% compared to 16% in the placebo group at the dose of 80 mg 4 weekly. After 6 months of 80 mg every 4 weeks, reductions in LDL-C, HDL-C, VLDL-C, non-HDL-C, and apo B of 7, 18, 47, 21, and 9%, respectively, were shown [4••, 37•]. Interestingly, there was no significant change in Lp(a) levels (3%) in this study [4••]. Treatment with vupanorsen in the phase I and early phase II studies was not associated with serious adverse events, with most adverse events being only mild to moderate and mainly related to injection site reactions [4••]. However, the clinical development of vupanorsen was discontinued on 31 January 2022, when results of the TRANSLATE-TIMI 70 trial, a phase IIb dose-escalating trial, designed to assess the effect on non-HDL-C levels, showed a disappointing placebo-adjusted reduction in non-HDL-C of 26.5% at 24 weeks. Reductions in LDL-C and apo B were only 7.9 and 8.5% respectively, even with the most effective dose of 160 mg given every 2 weeks. Furthermore, safety concerns included marked elevations in liver enzymes, with 39% of participants having elevations in ALT or AST > 3 the upper limit of normal with the 160 mg fortnightly dosing [45•]. Furthermore, a dose-dependent change in the fraction of hepatic fat, with a 76% relative increase from baseline, was observed in those on the 160-mg, 2-weekly dose [45•].
Small interfering RNA (siRNA)—ARO-ANG3
In contrast to ASO, siRNA therapy makes use of double-stranded DNA (dsDNA) and is recycled within the RNA-induced silencing complex with a resultant prolonged inhibition of ANGPTL3 mRNA and reduced hepatic synthesis of ANGPTL3, thus allowing for less frequent dosing as compared to other gene silencing approaches such as ASOs [37•]. ARO-ANG3 is a GalNAc-conjugated siRNA directed against ANGPTL3 mRNA.
Results from the phase I study in healthy volunteers with a fasting TG > 100 mg/dL (> 1.1 mmol/L) and LDL-C > 70 mg/dL (> 1.8 mmol/L), who received a single ascending dose of ARO-ANG3 subcutaneously (35–300 mg), showed mean reductions in serum TG, LDL-C, HDL-C, and VLDL-C of 52, 8, 16, and 52%, respectively, at 16 weeks with the 300-mg subcutaneous dose [37•, 46, 47•]. Participants of the multi-dose component of this phase I study received ARO-ANG3 at day 1 and day 29 at a dose of 100–300 mg subcutaneously. The mean nadir for ANGPTL3 reduction occurred 2 weeks after the second dose (− 83 to − 93%), with an observed mean decrease in TG and LDL-C of 67 and 36%, respectively, with the 300 mg subcutaneous dose after 16 weeks. A mean reduction in HDL-C and VLDL-C of 37 and 65% retrospectively was also shown [37•, 46, 47•].
Other interventional cohorts within this ongoing study include participants with HeFH as well as those without FH but with an LDL-C > 70 mg/dL (> 1.8 mmol/L) despite treatment with a statin with or without a PCSK9 inhibitor [37•, 46, 47•]. Preliminary results in the non-FH participants showed a reduction in TG and LDL-C of 29 and 28% at 16 weeks with the 200-mg dose [36, 45•, 46]. In those with HeFH, randomized to receive either of the multiple ascending doses of ARO-ANG3 (100, 200, 300 mg), mean ANGPTL3 levels were significantly reduced between 62 and 92% in a dose-dependent manner. A consistent reduction in TG (25–43%) and LDL-C (23–37%) was seen at all doses, with a reduction in TG, LDL-C, HDL-C, and VLDL-C at 16 weeks of 12, 35, 42, and 15% with the 300 mg dose [37•, 46, 47•]. These results have shown that the siRNA, ARO-ANG3, results in a safe and tolerable, dose-dependent reduction in ANGPTL3 and atherogenic lipoproteins, and thus may be a potential new therapy for addressing residual ASCVD risk in patients with dyslipidemia.
Future Therapeutic Strategies
ANGPTL3 inhibition is a promising new strategy for treating atherogenic dyslipidemia. However, the global implementation of treatment with anti-ANGPTL3 antibodies, ASO, or siRNAs is limited due to the economic burden with prolonged therapy required for these targeted agents. Thus, more cost-effective therapeutic options also need to be explored.
Gene Editing—CRISPR-Cas9 Technology
Clustered regularly interspaced palindromic repeats-associated protein 9 (CRISPR/Cas9)–mediated genome editing has emerged as a promising technology and is certainly a favorable future alternative to current long-term treatment options for hyperlipidemia [48]. The CRISPR/Cas9 system institutes a double-strand break at desired sites for genome editing. Repair of the excised DNA leads to insertions and/or deletions being introduced into the genome which results in frameshift mutations, effectively “knocking out” the gene [48].
Since LOF mutations in both PCSK9 and ANGPTL3 genes have been found in apparently healthy human subjects, these genes have been considered potential candidates for editing. In mouse models, the inactivation of PCSK9 demonstrated a 35–40% reduction in TC [49]. More recently, the base editing of both PCSK9 alleles led to a 60% reduction in LDL-C in primates [49, 50•]. Transient, mild elevations in liver enzymes were seen and off-target editing occurred at one site in primate subjects [50•].
Base editing of ANGPTL3 in a mouse model of HoFH led to a 56% reduction in TG and a 51% reduction in TC [51].
Effective delivery systems for CRISPR-Cas9 tools are still needed to ensure specific targeting. Off-target effects have been seen in mouse models of atherosclerosis, so further studies are needed to establish safety before these newer therapies can be used in humans.
Vaccination against ANGPTL3
Vaccine development is well underway for the management and prevention of several chronic, non-communicable diseases such as hypertension, Alzheimer’s disease, obesity, and cancer [52, 53]. The aim of therapeutic vaccination would be to induce a sufficient antibody response in order to neutralize a specific protein, but without eliciting a T-cell response, which could lead to tissue damage or even have life-threatening consequences [53,54,55].
An anti-PCSK9 peptide vaccine in a HeFH murine model (Ldlr+/−) led to a 50% reduction in LDL-C, an effect which was sustained for up to 1 year [55]. Vaccination against PCSK9 by means of a virus-like particle (VLP) vaccine achieved a modest 10–15% reduction in rhesus macaques, an effect which was subsequently amplified by concomitant statin use [56]. Self-antigens attached to the surface of virus-like particles (VLPs) between 25 and 100 nm in diameter are displayed in a dense, repetitive manner to overcome B-cell tolerance and induce a robust neutralizing autoantibody response [53, 57].
Proof-of-concept studies for vaccination against ANGPTL3 have also shown some success in murine models [57]. Reductions in circulating ANGPTL3 levels, non-fasting TG, and VLDL together with modest reductions in LDL-C and HDL-C were seen in both a dyslipidemic obese mouse model (ob/ob mice) and an FH mouse model vaccinated against ANGPTL3 [58•]. Effects of vaccination tended to wane, however, after 30 weeks [58•].
There are several reasons why vaccination is an attractive treatment approach. MAb have the potential to induce neutralizing anti-mAb antibodies by the host rendering the drug less effective. Vaccinations will likely require less frequent dosing as compared to mAb which have a relatively short in vivo half-life [58•]. Vaccines are generally more cost-effective than mAb which should lead to greater accessibility, particularly among lower-income countries [58•].
No vaccines against ANGPTL3 have, as yet, been reported in humans. Further studies are needed to obtain a more durable vaccine effect which would ensure cost-effectiveness. The potentially harmful auto-reactive T-cell responses also remain a concern.
ANGPTL3 as a Target for Triglyceride and LDL-C Reduction
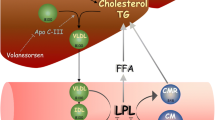
Strategies to reduce residual ASCVD risk have recently been focused on lowering TG levels. ANGPTL3 and apoC-III are critical in TG metabolism, with genetic evidence and clinical studies linking both these lipoproteins with increased ASCVD risk [59•]. As evidenced by the data presented in this review, ANGPTL3 is a novel therapeutic target for reducing hypertriglyceridemia as well as for lowering LDL-C further [7•]. Data from the APPROACH and COMPASS studies, evaluating the use of volanesorsen, an apoC-III inhibitor, showed up to a 77% reduction in TG; however, an increase in LDL-C was observed. The reduction in TG with volanesorsen is independent of the genetic variant affecting LPL activity, thus implying that volanersorsen also works via an LPL-independent mechanism. Studies suggest that ANGPTL3 and apoC-III inhibitors are equally potent in reducing TG and TRL levels across a wide range of concentrations; however, greater efficacy in reduction in these lipoproteins in those with severe hypertriglyceridemia may occur with apoC-III inhibition [7•]. Inhibition of ANGPTL3 has the benefit of greater LDL-C lowering and would thus be indicated in FH patients and those with mixed dyslipidemia that are not at target LDL-C levels despite best combination LLT (Fig. 1) [7•].

Strategies to reduce residual CV risk have recently been focused on lowering TG levels. ANGPTL3 inhibitors have the benefit of reducing both LDL-C and TGs, and are thus indicated in both familial and mixed hypercholesterolemia, whereby target LDL-C is not achieved despite optimal LLT. Both ANGPTL3 and APOC inhibition are equally effective in reducing TG levels; however, there may be greater efficacy of TG reduction with the APOC-III inhibitors. Inhibition of ANGPTL3 has the benefit of greater LDL-C lowering and would thus be indicated in FH patients and those with mixed dyslipidemia. Adapted by permission from Springer Nature from: Ward NC et al. BioDrugs. 2022;36(2):121–135) [7•]. ANGPTL3, angiopoietin-like 3; APOC-III, apolipoprotein C-III; FCS, familial chylomicronemia syndrome; HeFH, heterozygous familial hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia; LDL-C, low-density lipoprotein cholesterol; HC, hypercholesterolemia; LLT, lipid-lowering therapy; TG, triglycerides
Conclusion
The advances made with novel medical and biotechnological lipid-lowering therapeutic modalities have provided an opportunity to address not only a further reduction in LDL-C but also TRLs. With the dawn of a new era of LLT, the focus should be on targeted therapy by initiating early combination therapy in patients with high ASCVD risk. This can be successfully achieved with the rapidly growing armamentarium of LLT available and allows for a reduction in lipoproteins beyond LDL-C. TRLs or remnants have been shown to contribute independently to CV risk, and ANGPTL3 inhibition has the benefit of reducing both LDL-C and TRLs. Hence, ANGPTL3 inhibitors have the potential to reduce residual ASCVD risk in a broad range of dyslipidemic subjects including those with HoFH, HeFH, as well as refractory mixed dyslipidemia.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Musunuru K, Kathiresan S. Surprises from genetic analyses of lipid risk factors for atherosclerosis. Circ Res. 2016;118(4):579–85. https://doi.org/10.1161/CIRCRESAHA.115.306398.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Cholesterol Treatment Trialists’ (CTT) Collaborators Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet. 2005;366(9493):1267–78. https://doi.org/10.1016/S0140-6736(05)67394-1.
Nichols GA, Philip S, Reynolds K, Granowitz CB, Fazio S. Increased cardiovascular risk in hypertriglyceridemia patients with statin-controlled LDL cholesterol. J Clin Endocrinol Metab. 2018;103(8):3019–27. https://doi.org/10.1210/jc.2018-00470.
•• Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, Hurh E, Kingsbury J, Bartlett VJ, Vupanorsen Study Investigators, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridemia. Eur Heart J. 2020;41(40):3936–45. https://doi.org/10.1093/eurheartj/ehaa689. (Phase 2 study in patients with hypertriglyceridemia, diabetes and hepatic steatosis showed an improvement in the lipid profile with the ASO, Vupanorsen, compared to placebo.)
Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118(4):547–63. https://doi.org/10.1161/CIRCRESAHA.115.306249.
• Ginsberg HN, Packard CJ, Chapman MJ, Borén J, Aguilar-Salinas CA, Averna M, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies–a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42(47):4791–806. https://doi.org/10.1093/eurheartj/ehab551. (Consensus statement providing an updated therapeutic paradigm of targeting TRL and their remnants, with the aim of reducing the risk of ASCVD.)
• Ward NC, Chan DC, Watts GF. A tale of two new targets for hypertriglyceridemia: which choice of therapy? BioDrugs. 2022;36(2):121–35. https://doi.org/10.1007/s40259-022-00520-2. (A review of the etiology and treatment of hypertriglyceridemia, with a focus on the therapeutic role of ANGPTL3 and apoC-III inhibition.)
Young SG, Fong LG, Beigneux AP, Allan CM, He C, et al. GPIHBP1 and lipoprotein lipase, partners in plasma triglyceride metabolism. Cell Metab. 2019;30(1):51–65. https://doi.org/10.1016/j.cmet.2019.05.023.
Davies BS, Beigneux AP, Barnes RH 2nd, Tu Y, Gin P, Weinstein MM, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12(1):42–52. https://doi.org/10.1016/j.cmet.2010.04.016.
Adachi H, Fujiwara Y, Kondo T, Nishikawa T, Ogawa R, Matsumura T, et al. Angptl 4 deficiency improves lipid metabolism, suppresses foam cell formation and protects against atherosclerosis. Biochem Biophys Res Commun. 2009;379(4):806–11. https://doi.org/10.1016/j.bbrc.2008.12.018.
Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277(37):33742–8. https://doi.org/10.1074/jbc.M203215200.
Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13(12):731–9. https://doi.org/10.1038/nrendo.2017.119.
Ono M, Shimizugawa T, Shimamura M, Yoshida K, Noji-Sakikawa C, Ando Y, et al. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J Biol Chem. 2003;278(43):41804–9. https://doi.org/10.1074/jbc.M302861200.
• Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res. 2020;61(9):1271–86. https://doi.org/10.1194/jlr.RA120000888. (Research showing that ANGPTL3 inhibition promotes VLDL processing and clearance via an EL-dependent mechanism.)
Conklin D, Gilbertson D, Taft DW, Maurer MF, Whitmore TE, Smith DL, et al. Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics. 1999;62(3):477–82. https://doi.org/10.1006/geno.1999.6041.
Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30(2):151–7. https://doi.org/10.1038/ng814.
Essalmani R, Susan-Resiga D, Chamberland A, Asselin MC, Canuel M, Constam D, et al. Furin is the primary in vivo convertase of angiopoietin-like 3 and endothelial lipase in hepatocytes. J Biol Chem. 2013;288(37):26410–8. https://doi.org/10.1074/jbc.M113.501304.
Li X, Zhang Y, Zhang M, Wang Y. GALNT2 regulates ANGPTL3 cleavage in cells and in vivo of mice. Sci Rep. 2020;10(1):16168. https://doi.org/10.1038/s41598-020-73388-3.
Sylvers-Davie KL, Davies BSJ. Regulation of lipoprotein metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am J Physiol Endocrinol Metab. 2021;321(4):E493–508. https://doi.org/10.1152/ajpendo.00195.2021.
Quagliarini F, Wang Y, Kozlitina J, Grishin NV, Hyde R, Boerwinkle E, et al. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci U S A. 2012;109(48):19751–6. https://doi.org/10.1073/pnas.1217552109.
• Jin N, Matter WF, Michael LF, Qian Y, Gheyi T, Cano L, et al. The angiopoietin-like protein 3 and 8 complex interact with lipoprotein lipase and induces LPL cleavage. ACS Chem Biol. 2021;16(3):457–62. https://doi.org/10.1021/acschembio.0c00954. (This study demonstrated the mechanism by which ANGPTL3 inhibits LPL.)
Chen YQ, Pottant TG, Siegel RW, Ehsani M, Qian Y-W, Roell WC, Konrad RJ. Angiopoietin-like protein 4(E40K) ANGPTL4/8 complex have reduced, temperature-dependent LPL-inhibitory activity compared to ANGPTL4. Biochem Biophys Res Commun. 2021;534:498–503.
Fazio S, Sidoli A, Vivenzio A, Maietta A, Giampaoli S, Menotti A, et al. A form of familial hypobetalipoproteinaemia not due to a mutation in the apolipoprotein B gene. J Intern Med. 1991;229(1):41–7. https://doi.org/10.1111/j.1365-2796.1991.tb00304.x.
Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363(23):2220–7. https://doi.org/10.1056/NEJMoa1002926.
Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377(3):211–21. https://doi.org/10.1056/NEJMoa1612790.
Minicocci I, Santini S, Cantisani V, Stitziel N, Kathiresan S, Arroyo JA, Martí G, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54(12):3481–90. https://doi.org/10.1194/jlr.P039875.
Xu Y-X, Redon V, Yu H, Querbes W, Pirruccello J, Liebow A, Deik A, Trindade K, Wang X, Musunuru K, Clish CB, Cowan C, Fizgerald K, Rader D, Kathiresan S. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoproteins. Atherosclerosis. 2018;268:196–206. https://doi.org/10.1016/j.atherosclerosis.2017.08.031.
Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119(1):70–9. https://doi.org/10.1172/JCI37118.
Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, PROMIS and Myocardial Infarction Genetics Consortium Investigators, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69(16):2054–63. https://doi.org/10.1016/j.jacc.2017.02.030.
Chen PY, Gao WY, Liou JW, Lin CY, Wu MJ, Yen JH. Angiopoietin-like protein 3 (ANGPTL3) modulates lipoprotein metabolism and dyslipidemia. Int J Mol Sci. 2021;22(14):7310. https://doi.org/10.3390/ijms22147310.
Nidhina Haridas PA, Soronen J, Sädevirta S, Mysore R, Quagliarini F, Pasternack A, et al. Regulation of angiopoietin-like proteins (ANGPTLs) 3 and 8 by insulin. J Clin Endocrinol Metab. 2015;100(10):E1299–307. https://doi.org/10.1210/jc.2015-1254.
Shimamura M, Matsuda M, Ando Y, Koishi R, Yasumo H, Furukawa H, et al. Leptin and insulin down-regulate angiopoietin-like protein 3, a plasma triglyceride-increasing factor. Biochem Biophys Res Commun. 2004;322(3):1080–5. https://doi.org/10.1016/j.bbrc.2004.08.024.
Pramfalk C, Parini P, Gustafsson U, Sahlin S, Eriksson M. Effects of high-dose statin on the human hepatic expression of genes involved in carbohydrate and triglyceride metabolism. J Intern Med. 2011;269(3):333–9. https://doi.org/10.1111/j.1365-2796.2010.02305.x.
• Reeskamp LF, Tromp TR, Huijgen R, Stroes ESG, Hovingh GK, Grefhorst A. Statin therapy reduces plasma angiopoietin-like 3 (ANGPTL3) concentrations in hypercholesterolemic patients via reduced liver X receptor (LXR) activation. Atherosclerosis. 2020;315:68–75. https://doi.org/10.1016/j.atherosclerosis.2020.11.013. (This study showed that statins reduce ANGPTL3 levels by 15%.)
Laufs U, Parhofer KG, Ginsberg HN, Hegele RA. Clinical review on triglycerides. Eur Heart J. 2020;41(1):99–109c. https://doi.org/10.1093/eurheartj/ehz785.
Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA. 2019;321(4):364–73.
• Watts GF, Raal FJ, Chan DC. Transcriptomic therapy for dyslipidemias utilizing nucleic acids targeted at ANGPTL3. Future Cardiol. 2022;18(2):143–53. https://doi.org/10.2217/fca-2021-0096. (Review of the functional role of ANGPTL3 in lipid metabolism and the rationale for the development of targeted nucleic acid therapies to ANGPTL3, supported by clinical trial data.)
Kersten S. ANGPTL3 as therapeutic target. Curr Opin Lipidol. 2021;32(6):335–41. https://doi.org/10.1097/MOL.0000000000000789.
Gusarova V, Alexa CA, Wang Y, Rafique A, Kim JH, Buckler D, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res. 2015;56(7):1308–17. https://doi.org/10.1194/jlr.M054890.
•• Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, ELIPSE HoFH Investigators, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383(8):711–20. https://doi.org/10.1056/NEJMoa2004215. (ELIPSE HoFH study, studying the use of evinacumab, a monoclonal antibody to ANGPTL3, showed up to a 50% reduction in LDL-C in HoFH patients, even in those with null-null LDLR receptor mutations.)
Gaudet D, Gipe DA, Pordy R, Ahmad Z, Cuchel M, Shah PK, et al. ANGPTL3 inhibition in homozygous familial hypercholesterolemia. N Engl J Med. 2017;377(3):296–7. https://doi.org/10.1056/NEJMc1705994.
• Raal FJ, Rosenson RS, Reeskamp LF, Kastelein JJ, Rubba P, Duell B, et al. The longer-term efficacy of evinacumab in patients with homozygous familial hypercholesterolemia. Circulation. 2021;144:A12066. https://doi.org/10.1161/circ.144.suppl_1.12066. (Post hoc analysis of the ELIPSE HoFH study showed long-term efficacy of evinacumab, whereby 32% achieved an LDL-C <70 mg/dL (<1.8 mmol/L).)
•• Rosenson RS, Burgess LJ, Ebenbichler CF, Baum SJ, Stroes ESG, Ali S, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383(24):2307–19. https://doi.org/10.1056/NEJMoa2031049. (Evinacumab reduced LDL-C by more than 50% in HeFH and non-FH patients on maximal lipid-lowering therapy.)
Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377(3):222–32. https://doi.org/10.1056/NEJMoa1701329.
• Bergmark BA, Marston NA, Bramson CR, Curto M, Ramos V, Jevne A, Kuder JF, TRANSLATE-TIMI 70 Investigators, et al. Effect of vupanorsen on non-high-density lipoprotein cholesterol levels in statin-treated patients with elevated cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145(18):1377–86. https://doi.org/10.1161/CIRCULATIONAHA.122.059266. (Vupanorsen, at escalating doses, in adults with hyperlipidemia, showed a modest reduction in LDL-C, with a dose-dependent increase in liver enzymes and hepatic fat.)
Watts GF, Schwabe C, Scott R, Gladding P, Sullivan DR, Baker J, et al. RNA interference targeting hepatic & angiopoietin-like protein 3 results in prolonged reductions in plasma triglycerides and LDL-C in human subjects. Circulation. 2019;140:e987–8.
• Watts GF, Schwabe C, Scott R, Gladding P, Sullivan D, Baker J, et al. RNAi inhibition of angiopoietin-like protein 3 (ANGPTL3) with ARO-ANG3 mimics the lipid and lipoprotein profile of familial combined hypolipidemia. Eur Heart J. 2020;41(2):ehaa946.3331. https://doi.org/10.1093/ehjci/ehaa946.3331. (Sequential doses of an siRNA against ANGPTL3 administered to healthy volunteers dropped ANGPTL3 levels by more than 80% leading to a lowering of TG > 60% and LDL-C >45%.)
Siew WS, Tang YQ, Kong CK, Goh BH, Zacchigna S, Dua K, et al. Harnessing the potential of CRISPR/Cas in atherosclerosis: disease modeling and therapeutic applications. Int J Mol Sci. 2021;22(16):8422. https://doi.org/10.3390/ijms22168422.
Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115(5):488–92. https://doi.org/10.1161/CIRCRESAHA.115.304351.
• Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593(7859):429–34. https://doi.org/10.1038/s41586-021-03534-y. (Proof-of-concept study in nonhuman primates showing that CRISPR base editing lowers PCSK9 and LDL-C by 90% and 60% respectively.)
Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation. 2018;137(9):975–7. https://doi.org/10.1161/CIRCULATIONAHA.117.031335.
Bachmann MF, Whitehead P. Active immunotherapy for chronic diseases. Vaccine. 2013;31(14):1777–84. https://doi.org/10.1016/j.vaccine.2013.02.001.
Jennings GT, Bachmann MF. Immunodrugs: therapeutic VLP-based vaccines for chronic diseases. Annu Rev Pharmacol Toxicol. 2009;49:303–26. https://doi.org/10.1146/annurev-pharmtox-061008-103129.
Sahebkar A, Momtazi-Borojeni AA, Banach M. PCSK9 vaccine: so near, yet so far! Eur Heart J. 2021;42(39):4007–10. https://doi.org/10.1093/eurheartj/ehab299.
Galabova G, Brunner S, Winsauer G, Juno C, Wanko B, Mairhofer A, et al. Peptide-based anti-PCSK9 vaccines – an approach for long-term LDLc management. PLoS ONE. 2014;9(12):e114469. https://doi.org/10.1371/journal.pone.0114469.
Crossey E, Amar MJA, Sampson M, Peabody J, Schiller JT, Chackerian B, et al. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine. 2015;33(43):5747–55. https://doi.org/10.1016/j.vaccine.2015.09.044.
Fowler A, Sampson M, Remaley AT, Chackerian B. A VLP-based vaccine targeting ANGPTL3 lowers plasma triglycerides in mice. Vaccine. 2021;39(40):5780–6. https://doi.org/10.1016/j.vaccine.2021.08.077.
• Fukami H, Morinaga J, Nakagami H, Hayashi H, Okadome Y, Matsunaga E, et al. Vaccine targeting ANGPTL3 ameliorates dyslipidemia and associated diseases in mouse models of obese dyslipidemia and familial hypercholesterolemia. Cell Rep Med. 2021;2(11):100446. https://doi.org/10.1016/j.xcrm.2021.100446. (A peptide vaccine against ANGPTL3 in mouse models of dyslipidemia durably lowered TG and LDL-C for 30 weeks.)
• Reeskamp LF, Tromp TR, Stroes ESG. The next generation of triglyceride-lowering drugs: will reducing apolipoprotein C-III or angiopoietin like protein 3 reduce cardiovascular disease? Curr Opin Lipidol. 2020;31(3):140–6. https://doi.org/10.1097/MOL.0000000000000679. (Review of the role of apoC-III and ANGPTL3 in triglyceride metabolism.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
FJR has received research grants, honoraria, or consulting fees for professional input and/or delivered lectures from Sanofi-Aventis, Regeneron, Amgen, Novartis, and LIB Therapeutics. FM and BSM have no disclosures.
Human and Animal Rights and Informed Consent
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Human Research Ethics Committee, University of the Witwatersrand, Johannesburg, South Africa; Reference number: 180214) and with the 1964 Helsinki declaration and its later amendments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Genetics and Genomics.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohamed, F., Mansfield, B.S. & Raal, F.J. ANGPTL3 as a Drug Target in Hyperlipidemia and Atherosclerosis. Curr Atheroscler Rep 24, 959–967 (2022). https://doi.org/10.1007/s11883-022-01071-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-022-01071-1