
Abstract
Purpose of Review
Despite the important progress in identifying high-risk atherosclerotic plaques, many key elements are elusive. Advanced imaging modalities provide valuable information about the anatomic and functional plaque characteristics and underscore the presence of multiple plaque morphologies. However, how the heterogeneity of atherosclerotic plaque can alter our current understanding of coronary artery disease is not fully understood.
Recent Findings
Along the length of an individual plaque, the morphology patterns display marked heterogeneity. Contrary to previous beliefs, plaque morphology is also highly dynamic over time, with the vast majority of high-risk plaques becoming quiescent and mild plaques becoming severely obstructive in a short period of time. Endothelial shear stress, a local hemodynamic factor known for its critical effects in plaque initiation and progression, also displays longitudinal heterogeneity contributing to the arterial wall response in all time points.
Summary
Risk stratification of plaques based on the morphological characteristics at one region of the plaque, usually the minimal lumen diameter, and at one point in time may be misleading. The evaluation of both morphological and hemodynamic characteristics along the length of a plaque will improve the risk assessment of individual plaques.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding the pathophysiologic mechanisms behind coronary atherosclerosis is of utmost importance for preventing and efficiently treating coronary disease complications. For decades, the only available data about the morphologic characteristics of lesions came from post-mortem studies. The advent of new imaging techniques enabled the in vivo evaluation not only of anatomical but also of functional characteristics of lesions and provided valuable information about the natural history of atherosclerosis [1–4]. The detailed assessment of individual plaques revealed considerable heterogeneity of the morphology and the local hemodynamic milieu both along the length of each plaque and over time [5••, 6, 7•, 8]. The clinical interpretation of these new data may enrich our knowledge on the atherosclerotic mechanisms and improve the short- and long-term risk stratification of patients.
Mechanisms of Atherosclerotic Plaque Formation
The healthy endothelium forms a barrier between blood flow and the arterial wall, senses the different types of biomechanical forces, and regulates numerous mechanisms of vascular homeostasis such as vascular tone, transportation of molecules, coagulation state and fibrinolysis. In atheroprone regions, and in the presence of coronary disease risk factors, vascular smooth muscle cells (VSMCs) migrate into the intima, proliferate, and elaborate molecules into the extracellular matrix (ECM) environment. Interaction of low-density lipoprotein (LDL) particles with ECM components, especially proteoglycans, facilitates the sub-endothelial retention of LDL long enough for their oxidation by reactive oxygen species (ROS). This reaction further augments endothelial dysfunction and triggers inflammatory pathways that result in monocyte recruitment, internalization, and differentiation into macrophages [9].
A continuous feedback between activated macrophages, dysfunctional endothelial cells, VSMCs, and LDL particles potentiates and aggravates the atherosclerotic process that gradually leads to plaque formation. Accumulated extracellular lipoproteins in the proteoglycan-collagen enriched ECM form the initial progressive atherosclerotic lesion, pathologic intimal thickening (PIT). The infiltration of macrophages within the lipid pool (accumulated lipoproteins) and their subsequent apoptosis result in the development of the lipid-rich necrotic core that, combined with a collagen fibrous cap, constitute the advanced lesion, a fibroatheroma [10].
Neovascularization, or formation of new vessels in the arterial wall, contributes to the progression of atherosclerosis, and is mainly triggered by the hypoxia attributed to intimal thickening and increased plaque size. The formation of new vessels provides the atherosclerotic lesion with oxygen and nutrient supplies and thus promotes plaque growth by reducing the limitation of diffusion distance; however, neovessels also supply inflammatory cells and pro-atherosclerotic molecular mediators to the plaque from the perivascular tissue. These newly formed vessels are also prone to rupture and hemorrhage due to compromised structural integrity related to a discontinuous endothelial layer and absence of smooth muscle cells [11, 12]. Intraplaque hemorrhage may also originate from subclinical plaque rupture or endothelial and fibrous cap injury [13]. This intraplaque hemorrhage constitutes a major contributing factor exacerbating plaque progression and instability since the free lipids from the red blood cell membranes and the released hemoglobin and iron lead to local fibrosis and scarring [14, 15]. Repeated cycles of subclinical plaque rupture and subsequent intraplaque hemorrhage may lead to progressively accelerated luminal obstruction clinically manifesting as worsening angina.
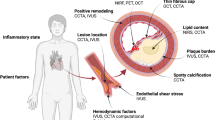
Arterial remodeling is another major determinant of plaque morphology and possible progression. Remodeling is an adaptive mechanism which reflects the ability of the vascular wall to adjust its dimensions in response to plaque growth or flow alterations. Positive or expansive remodeling denotes an increase in arterial dimensions, while negative or constrictive remodeling signifies that the arterial size decreases [16]. Compensatory expansive remodeling, as first highlighted by Glagov and colleagues in 1987 [17], is a process whereby the outer arterial wall enlarges to accommodate the plaque present within the arterial wall so that the lumen dimensions remain unchanged and distal blood flow is preserved. Excessive expansive remodeling, in contrast, is associated with highly inflamed atheroma, where the activated leukocytes elaborate plaque-degrading collagenases and elastases which disrupt the plaque structural integrity and lead to a focal excessive enlargement of the lumen and the arterial wall. These areas of excessive expansive remodeling have been consistently associated with high-risk plaque features leading to acute plaque rupture and acute coronary syndromes [18].
Stages of Atherosclerotic Plaque Progression
The earliest type of atherosclerotic lesion encountered in the coronary arteries is the PIT which is formed of smooth muscle cells arranged in layers in an ECM composed of proteoglycans and collagen fibers that overlie a lipid pool rich in low density lipoproteins, with variable infiltration of macrophages, presence of cholesterol clefts, and microcalcifications [19]. Fibroatheromas are composed of a necrotic core covered by a thick fibrous cap of smooth muscle cells and matrix of proteoglycans and collagen fiber types I and III [20]. Thin-cap fibroatheromas (TCFAs) are typically regarded as the precursor lesions responsible for adverse coronary events [7•] and are characterized by a large lipid and necrotic core, a thin fibrous cap and intense inflammatory cell infiltration [10, 21, 22]. The adverse clinical natural history manifestations of local atherosclerotic plaque may be triggered by a variety of pathobiological processes, such as endothelial denudation, fibrous cap disruption, intraplaque hemorrhage, endothelial dysfunction, and expansive remodeling [23–25].
Dynamic Nature of Plaque Progression and Natural History
The progression of an atherosclerotic plaque through the stages noted above suggests that atherosclerosis progresses inexorably in a linear and monotonic manner. In sharp contradistinction, however, recent evidence from animal and human studies indicates that there is a remarkably dynamic and heterogeneous evolution of each plaque over time.
Arterial remodeling of individual plaques seems to be very dynamic over time, according to experimental data from animal studies. A considerable percentage (∼75 %) of ostensibly high-risk TCFAs in human arteries may heal and become quiescent during the natural course of atherosclerosis, possibly because of arterial remodeling that may reduce the local pro-inflammatory low endothelial shear stress (ESS) environment, underscoring the complexity of pathophysiologic mechanisms leading to clinical events (Fig. 1) [5••, 26••, 27]. Furthermore, it has recently become clear that the phenotypic manifestations of atherosclerosis are not uniform even along the length of an individual plaque, but are very heterogeneous with a variety of plaque morphologies, remodeling characteristics, and local hemodynamic environments along their length, and the longer the plaque length the more heterogeneous the various plaque characteristics along that plaque (Fig. 2) [28].

Dynamic natural history of local plaque characteristics in individual porcine coronary plaques. Intravascular ultrasound-based 3D artery reconstruction was serially performed at 5 consecutive time points in vivo in 5 diabetic, hypercholesterolemic pigs. ESS and vascular remodeling were assessed along the course of each of the 3 coronary arteries. a Local arterial remodeling characteristics. A dynamic and heterogeneous progress in arterial remodeling of individual lesions was noted over time. The majority of segments with compensatory remodeling remained with that remodeling pattern over time. Only a small minority of segments with either excessive expansive or constrictive remodeling at week 4 continued to exhibit the same remodeling pattern throughout their evolution (adapted from Reference [26••], with permission). b Local ESS characteristics. Local ESS in individual segments (n = 184) frequently changed from low ESS (<1.2 Pa; lower row) to higher ESS (intermediate/high, ≥1.2 Pa; upper row) between consecutive time points. The black portion of the pie charts at each time point represents the proportion of segments with low ESS at the immediately preceding time point; the white portion represents the proportion of segments with higher ESS at the preceding time point (reprinted from Reference [27], with permission)

Heterogeneity of local plaque characteristics within individual human plaques. Coronary angiography and IVUS-derived 3D artery reconstruction was performed in 219 patients of the PREDICTION study, and 371 plaques were identified (adapted from Reference [28], with permission). a Arterial remodeling patterns per plaque. The number of arterial remodeling patterns in an individual plaque progressively increased as the plaque became longer. b. ESS patterns per plaque. The number of ESS patterns in an individual plaque progressively increased as the plaque became longer
The distribution of plaques in the coronary arteries is non-uniform, showing a predilection for the proximal parts of coronary arteries, the lateral walls of arterial bifurcations, inner parts of arterial curvatures, and upstream or downstream from luminal obstructions, whereas other areas remain relatively unaffected [29–31]. Furthermore, plaque morphology around the circumference of the arterial wall is typically very eccentric and heterogeneous. The rate of growth of each plaque is variable, and plaques at different stages of evolution typically co-exist within the same artery, reflecting the highly individual natural history of each plaque [26••].
The Pathogenesis of Plaque Heterogeneity
Local Intracoronary Hemodynamic Factors
Blood circulation through the coronary arteries and cardiac motion produces mechanical forces inflicted axially, radially, and circumferentially on the vessel wall. For computational purposes, these forces are better described as stresses, i.e., a force normalized by the area that the force is applied. ESS and tensile stress (TS) are the two stresses that critically modulate the atherogenic process and contribute to the regional and local heterogeneity of atherosclerosis [32–34]. Advances in computational techniques (computational fluid dynamics, finite element analysis) and intracoronary imaging modalities (intravascular ultrasound [IVUS], optical coherence tomography [OCT], coronary computed tomography angiography [CCTA]) allow for the in vivo estimation of hemodynamic forces [34, 35].
TS depends on hydrostatic (blood pressure), structural (lumen diameter, arterial wall thickness), and morphological (arterial wall composition) parameters. TS increases with higher arterial blood pressure, higher vessel lumen radius, and thinner arterial walls [36]. The composition and geometry of the arterial wall may alter TS values. According to experimental data, an increase in necrotic core size, a reduction in fibrous cap thickness, or the presence of microcalcification can increase TS [34, 37–40]. In addition, plaque regions with maximum lumen curvature such as the plaque shoulder are subjected to higher TS [37]. On the cellular level, TS affects VSCMs function including activation, migration, and apoptosis and, thus, contributes to atherosclerosis progression [41–44]. The distribution of TS along the length of a plaque and the site of maximum TS is of great interest as it correlates with the plaque rupture region [45].
ESS is the tangential force produced by the friction of the flowing blood to the endothelial surface and depends on blood viscosity and velocity. Low ESS (<1.5 Pa) provokes molecular, cellular, and vascular adaptations in areas prone to plaque development, leading to the initiation and progression of atherosclerosis [46]. The focal formation of lesions in specific predilection sites follows the different flow patterns created as blood circulates through the coronary tree. Arterial sites with curvatures, bifurcations, and upstream or downstream from obstructions exhibit disturbed local flow and low ESS in contrast to the laminar, undisturbed flow and physiologic (1.5–3 Pa) or high (>3.0 Pa) ESS in arterial segments that consistently exhibit a vasculoprotective phenotype. Endothelial mechanosensors sensitive to blood flow patterns and ESS stimuli can alter endothelial cell conformation and intracellular gene expression [47, 48]. Biomechanical stimuli act through intracellular mechanisms to activate a cascade of biochemical signals that modify gene transcription and expression through the anti-inflammatory transcription factor (TF) Kruppel-like factor 2 (KLF2) and the pro-inflammatory nuclear factor-κB (NF-κB) [49].
The structural and molecular responses of endothelial cells to a low or physiologic ESS milieu form an atheroprone or atheroprotective phenotype, respectively. Physiologic ESS regions are characterized by endothelial cells in a polygonal shape, elongated and aligned with laminar flow. On the other hand, in low ESS areas, cells are poorly aligned, and their structure is modified to a fusiform shape. The structural and orientation cell changes, combined with the accelerated endothelial cell turnover in low ESS sites, result in the widening of cell-to-cell junctions, which increases the permeability and facilitates the transmigration of molecules through the endothelium and into the intima [47, 50].
LDL accumulation in the sub-endothelium is a key step in the pathogenesis of atherosclerosis. In low ESS regions, increased endothelium permeability is also augmented by the upregulation of genes encoding for the LDL receptor of the endothelial membrane. Additionally, the prolonged residence time of the LDL particles close to the endothelium in low ESS regions works synergistically with the increased permeability towards internalization of LDL particles [46, 51, 52]. Hyperlipidemia is a precondition for the initiation of atherosclerosis, underscoring the important interaction between local and systemic factors. Recent findings supporting a cholesterol-dependent atherogenic threshold of low ESS further highlight the interplay between local and systemic variables [53]. The myriad effects and the balance of the pro-atherogenic and pro-inflammatory local low ESS are critical for the plaque both to develop and to progress.
Plaque Heterogeneity Along Individual Plaques
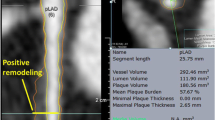
There is substantial heterogeneity in plaque composition and morphology within individual coronary atherosclerotic plaques [54]. An early IVUS study identified heterogeneity in plaque composition in nearly 9 out of 10 plaques, while the remodeling pattern was heterogeneous in about one fourth of plaques [6]. A recent study from the PREDICTION (Prediction of Progression of Coronary Artery Disease and Clinical Outcomes Using Vascular Profiling of Shear Stress and Wall Morphology) study detailed three-dimensional (3D) assessments of human coronary arterial and plaque morphology and confirmed the above findings, showing that a considerable proportion of plaques exhibit a combination of two or three remodeling patterns, as well as ESS profiles, concurrently along their length. Coronary plaque length is also highly variable ranging from 9 to 30 mm in various proportions [28]. The consequence of this plaque heterogeneity is that at any given point in time, different regions of a plaque can exhibit different stages of progression and development and, accordingly, different magnitudes of pathobiologic stimuli for further atherosclerotic progression or quiescence [28, 40, 55, 56]. These different risk profiles along the course of an individual plaque also change markedly over time. A simple risk assessment of an individual plaque at a single point in time based on the plaque imaging characteristics at the minimal lumen diameter, which is the usual clinical measure, may consequently seriously misrepresent the actual risk of that plaque. The actual morphological and risk complexity constitutes an important challenge for cardiovascular imaging modalities employed for the risk-stratification of CAD.
Plaque Heterogeneity Over Time
As a consequence of the local plaque heterogeneity, the local pathobiologic stimuli for plaque progression and the local adaptive or maladaptive arterial remodeling responses are highly dynamic over time [26••]. The extent of plaque progression or plaque healing will depend on the magnitude of both the stimulus for progression and the stimulus for healing and quiescence at that time. Clinical studies underscore that a considerable proportion of ostensibly high-risk plaques become quiescent over time [7•, 8].
An understanding of the actively ongoing vascular mechanisms and interactions along the plaque length, as well as the development and optimization of in-vivo imaging techniques to identify the magnitude and extent of these different processes, is essential for the early identification of coronary plaque regions more likely to exhibit a high-risk profile and thereby justify preemptive measures to avert any imminent adverse clinical sequelae.
Clinical Significance of Plaque Heterogeneity
Early identification of vulnerable plaques to enable prevention of major adverse cardiovascular events (MACE) is of critical importance. A number of recent studies investigating the presence of high-risk plaques, typically non-obstructing in nature, and the value of pre-emptive revascularization of these plaques have been controversial. The recent Preventive Angioplasty in Acute Myocardial Infarction (PRAMI) trial was the largest prospective study examining preemptive stenting of non-culprit plaques during primary PCI, with risk assessment based on angiography alone [57]. The favorable results of PRAMI trial in preventing MACE were, however, greeted with skepticism and concerns about the appropriate criteria for the selection of the lesions to be treated [58]. The limitations of coronary angiography in identifying the high-risk plaques are well known, and advanced imaging modalities that can focus on the arterial wall as well as the plaque, such as IVUS, OCT, near-infrared spectroscopy (NIRS), and CCTA, have far more potential to identify those plaques most likely to cause future MACE and therefore justify pre-emptive intervention [3, 4]. Magnetic resonance imaging (MRI) is also emerging as a promising modality in the field of coronary imaging and plaque characterization [59]. MRI as a non-invasive, radiation-free imaging modality would be an ideal candidate for screening in apparently healthy population. Results from animal studies encourage the wider use of MRI in human studies aiming to identify morphological and hemodynamic parameters of plaque vulnerability [60–62].
The Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) and PREDICTION studies were the largest invasive clinical natural history studies of coronary atherosclerosis. In PROSPECT, previously untreated coronary lesions associated with subsequent MACE (non-culprit lesions) were characterized at baseline by large plaque burden (PB) ≥70 %, small minimal luminal area (MLA) ≤4.0 mm2, and TCFA morphology [7•]. However, the vast majority of these ostensibly high-risk plaques became quiescent: <4 % of large PB lesions and <5 % of TCFA phenotype lesions actually caused MACE at follow-up. The PREDICTION study focused on the prognostic significance of both the plaque anatomy as well as the local pro-inflammatory ESS on the progression of individual plaques [8]. The positive predictive value to predict plaque progression treated with a percutaneous coronary intervention was 22 % based on plaque anatomy alone (large PB and small MLA), but rose to 41 % when the baseline pro-inflammatory low ESS was also included in the risk assessment. Adding radiofrequency IVUS-based tissue characteristics of large necrotic core to the risk assessment further increased the positive predictive value to 53 % while maintaining a high negative predictive value of 91 % if these predictors were absent [63•].
A critical synergy between pro-inflammatory local ESS and plaque anatomy, and the significance of plaque heterogeneity, to predict MACE was underscored by preliminary results from a recent post-hoc analysis of the PROSPECT study, which suggested that pro-inflammatory low ESS was essential for high-risk plaque anatomy to progress and lead to MACE in follow-up. MACE outcomes did not occur if the anatomic lesion did not exhibit local low ESS at baseline, regardless of PB, MLA, or lesion phenotype [64]. Furthermore, consistent with the significance of plaque heterogeneity as identified in the PREDICTION study, this post-hoc study demonstrated that non-culprit MACE lesions were more heterogeneous and significantly longer compared to non-culprit lesions without MACE and that the culprit low ESS responsible for MACE could be found anywhere along the length of the entire plaque [65].
The results from the post-hoc analysis of the PROSPECT study are in conflict with previously published data supporting a correlation between high ESS and plaque rupture. According to one study, high ESS values often co-localized with the plaque rupture site. Of note, the evaluation of ESS was performed after the plaque rupture [66]. Other results relate high ESS sites with increased strain values after 6-month follow-up, concluding that elevated ESS may contribute to a high-risk plaque profile [67]. Regardless of the discrepancies between reported results, all studies support the notion that interpreting plaque heterogeneity may significantly improve individual cardiac risk assessment, underscoring the need for large, natural history studies that will examine the morphological and hemodynamic characteristics of coronary atherosclerosis.
Conclusions
The identification of high-risk plaques, especially in humans, remains challenging. As technology advances and new imaging modalities become available, the complexity of a plaque’s anatomic and hemodynamic characteristics emerges as a promising risk predictor. Currently, several in-vivo imaging options are available which can characterize plaque anatomy and composition, i.e., coronary angiography, IVUS, radiofrequency-IVUS (virtual histology), OCT, and NIRS. Innovative new approaches can now identify the underlying pathophysiologic processes that are essential to plaque progression and destabilization. Such approaches are vascular profiling (with computational fluid dynamics) to assess local pro-inflammatory low ESS, characterization of the mechanical properties of plaque, and molecular imaging of inflammation. The combination of the above methods act synergistically to expand our insight concerning the natural history of atherosclerosis along the heterogeneous length of each individual plaque and hold enormous promise to identify the small subset of plaques that will continue to progress and ultimately lead to MACE. Studies assessing MLA characteristics alone have proven inadequate for accurate identification of vulnerable plaques. Evaluation of plaque morphology and ESS distribution along the length of the plaque may provide critical information that may alter the risk stratification of plaques and provide critical justification for pre-emptive interventions to avert adverse cardiac events.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Coskun AU, Yeghiazarians Y, Kinlay S, Clark ME, Ilegbusi OJ, Wahle A, et al. Reproducibility of coronary lumen, plaque, and vessel wall reconstruction and of endothelial shear stress measurements in vivo in humans. Catheter Cardiovasc Interv. 2003;60(1):67–78. doi:10.1002/ccd.10594.
Maehara A, Cristea E, Mintz GS, Lansky AJ, Dressler O, Biro S, et al. Definitions and methodology for the grayscale and radiofrequency intravascular ultrasound and coronary angiographic analyses. J Am Coll Cardiol Img. 2012;5(3 Suppl):S1–9. doi:10.1016/j.jcmg.2011.11.019.
Koskinas KC, Ughi GJ, Windecker S, Tearney GJ, Raber L. Intracoronary imaging of coronary atherosclerosis: validation for diagnosis, prognosis and treatment. Eur Heart J. 2016;37(6):524–35. doi:10.1093/eurheartj/ehv642.
Maurovich-Horvat P, Ferencik M, Voros S, Merkely B, Hoffmann U. Comprehensive plaque assessment by coronary CT angiography. Nat Rev Cardiol. 2014;11(7):390–402. doi:10.1038/nrcardio.2014.60.
•• Kubo T, Maehara A, Mintz GS, Doi H, Tsujita K, Choi SY, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55(15):1590–7. doi:10.1016/j.jacc.2009.07.078. An important natural history study that underscores the dynamic nature of coronary atherosclerotic plaques.
Box LC, Angiolillo DJ, Suzuki N, Box LA, Jiang J, Guzman L, et al. Heterogeneity of atherosclerotic plaque characteristics in human coronary artery disease: a three-dimensional intravascular ultrasound study. Catheter Cardiovasc Interv. 2007;70(3):349–56. doi:10.1002/ccd.21088.
• Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226–35. doi:10.1056/NEJMoa1002358. The largest clinical natural history study to examine the association of plaque tissue characteristics with the occurrence of adverse cardiac events.
Stone PH, Saito S, Takahashi S, Makita Y, Nakamura S, Kawasaki T, et al. Prediction of progression of coronary artery disease and clinical outcomes using vascular profiling of endothelial shear stress and arterial plaque characteristics: the PREDICTION Study. Circulation. 2012;126(2):172–81. doi:10.1161/CIRCULATIONAHA.112.096438.
Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. 2014;103(4):452–60. doi:10.1093/cvr/cvu171.
Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13–8. doi:10.1016/j.jacc.2005.10.065.
Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25(10):2054–61. doi:10.1161/01.ATV.0000178991.71605.18.
Fong GH. Potential contributions of intimal and plaque hypoxia to atherosclerosis. Curr Atheroscler Rep. 2015;17(6):510. doi:10.1007/s11883-015-0510-0.
Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103(7):934–40. doi:10.1161/01.CIR.103.7.934.
Xu J, Lu X, Shi GP. Vasa vasorum in atherosclerosis and clinical significance. Int J Mol Sci. 2015;16(5):11574–608. doi:10.3390/ijms160511574.
Nakashima Y, Chen YX, Kinukawa N, Sueishi K. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002;441(3):279–88. doi:10.1007/s00428-002-0605-1.
Feldman CL, Coskun AU, Yeghiazarians Y, Kinlay S, Wahle A, Olszewski ME, et al. Remodeling characteristics of minimally diseased coronary arteries are consistent along the length of the artery. Am J Cardiol. 2006;97(1):13–6. doi:10.1016/j.amjcard.2005.07.121.
Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371–5. doi:10.1056/NEJM198705283162204.
Varnava AM, Mills PG, Davies MJ. Relationship Between Coronary Artery Remodeling and Plaque Vulnerability. Circulation. 2002;105(8). doi:10.1161/hc0802.104327.
Kolodgie FD, Burke AP, Nakazawa G, Virmani R. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler Thromb Vasc Biol. 2007;27(5):986–9. doi:10.1161/ATVBAHA.0000258865.44774.41.
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262–75. doi:10.1161/01.ATV.20.5.1262.
Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336(18):1276–82. doi:10.1056/NEJM199705013361802.
Davies MJ. The composition of coronary-artery plaques. N Engl J Med. 1997;336(18):1312–4. doi:10.1056/NEJM199705013361809.
Fichtlscherer S, Breuer S, Zeiher AM. Prognostic value of systemic endothelial dysfunction in patients with acute coronary syndromes: further evidence for the existence of the “vulnerable” patient. Circulation. 2004;110(14):1926–32. doi:10.1161/01.CIR.0000143378.58099.8C.
Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349(24):2316–25. doi:10.1056/NEJMoa035655.
Schoenhagen P, Ziada KM, Kapadia SR, Crowe TD, Nissen SE, Tuzcu EM. Extent and direction of arterial remodeling in stable versus unstable coronary syndromes : an intravascular ultrasound study. Circulation. 2000;101(6):598–603. doi:10.1161/01.CIR.101.6.598.
•• Koskinas KC, Feldman CL, Chatzizisis YS, Coskun AU, Jonas M, Maynard C, et al. Natural history of experimental coronary atherosclerosis and vascular remodeling in relation to endothelial shear stress: a serial, in vivo intravascular ultrasound study. Circulation. 2010;121(19):2092–101. doi:10.1161/CIRCULATIONAHA.109.901678. An experimental natural history study that in addition to reporting plaque heterogeneity over time underlines the association of endothelial shear stress with the morphological variations of the plaque in time.
Koskinas KC, Sukhova GK, Baker AB, Papafaklis MI, Chatzizisis YS, Coskun AU, et al. Thin-capped atheromata with reduced collagen content in pigs develop in coronary arterial regions exposed to persistently low endothelial shear stress. Arterioscler Thromb Vasc Biol. 2013;33(7):1494–504. doi:10.1161/ATVBAHA.112.300827.
Antoniadis APPM, Takahashi S, Shishido K, Andreou I, Chatzizisis YS, Tsuda M, et al. Arterial remodeling and endothelial shear stress exhibit significant longitudinal heterogeneity along the length of coronary plaques. J Am Coll Cardiol Img. 2016;9(8):1007–9. doi:10.1016/j.jcmg.2016.04.003.
Asakura T, Karino T. Flow patterns and spatial distribution of atherosclerotic lesions in human coronary arteries. Circ Res. 1990;66(4):1045–66. doi:10.1161/01.RES.66.4.1045.
Giannoglou GD, Soulis JV, Farmakis TM, Farmakis DM, Louridas GE. Haemodynamic factors and the important role of local low static pressure in coronary wall thickening. Int J Cardiol. 2002;86(1):27–40. doi:10.1016/S0167-5273(02)00188-2.
Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. Jama. 1999;282(21):2035–42. doi:10.1001/jama.282.21.2035.
Richter Y, Edelman ER. Cardiology is flow. Circulation. 2006;113(23):2679–82. doi:10.1161/CIRCULATIONAHA.106.632687.
Gimbrone Jr MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–9. doi:10.1111/j.1749-6632.2000.tb06318.x. discussion 9–40.
Brown AJ, Teng Z, Evans PC, Gillard JH, Samady H, Bennett MR. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat Rev Cardiol. 2016;13(4):210–20. doi:10.1038/nrcardio.2015.203.
Holzapfel GA, Mulvihill JJ, Cunnane EM, Walsh MT. Computational approaches for analyzing the mechanics of atherosclerotic plaques: a review. J Biomech. 2014;47(4):859–69. doi:10.1016/j.jbiomech.2014.01.011.
Chatzizisis YS, Giannoglou GD. Coronary hemodynamics and atherosclerotic wall stiffness: a vicious cycle. Med Hypotheses. 2007;69(2):349–55. doi:10.1016/j.mehy.2006.11.053.
Teng Z, Sadat U, Li Z, Huang X, Zhu C, Young VE, et al. Arterial luminal curvature and fibrous-cap thickness affect critical stress conditions within atherosclerotic plaque: an in vivo MRI-based 2D finite-element study. Ann Biomed Eng. 2010;38(10):3096–101. doi:10.1007/s10439-010-0078-3.
Ohayon J, Finet G, Gharib AM, Herzka DA, Tracqui P, Heroux J, et al. Necrotic core thickness and positive arterial remodeling index: emergent biomechanical factors for evaluating the risk of plaque rupture. Am J Physiol Heart Circ Physiol. 2008;295(2):H717–27. doi:10.1152/ajpheart.00005.2008.
Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, et al. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303(5):H619–28. doi:10.1152/ajpheart.00036.2012.
Imoto K, Hiro T, Fujii T, Murashige A, Fukumoto Y, Hashimoto G, et al. Longitudinal structural determinants of atherosclerotic plaque vulnerability: a computational analysis of stress distribution using vessel models and three-dimensional intravascular ultrasound imaging. J Am Coll Cardiol. 2005;46(8):1507–15. doi:10.1016/j.jacc.2005.06.069.
Haga JH, Li YS, Chien S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech. 2007;40(5):947–60. doi:10.1016/j.jbiomech.2006.04.011.
Shyu KG. Cellular and molecular effects of mechanical stretch on vascular cells and cardiac myocytes. Clin Sci. 2009;116(5):377–89. doi:10.1042/CS20080163.
Rodriguez AI, Csanyi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, et al. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2015;35(2):430–8. doi:10.1161/ATVBAHA.114.304936.
Kwak BR, Back M, Bochaton-Piallat ML, Caligiuri G, Daemen MJ, Davies PF et al. Biomechanical factors in atherosclerosis: mechanisms and clinical implications. Eur Heart J. 2014;35(43):3013–20, 20a-20d. doi:10.1093/eurheartj/ehu353.
Thondapu V, Bourantas CV, Foin N, Jang IK, Serruys PW, Barlis P. Biomechanical stress in coronary atherosclerosis: emerging insights from computational modelling. Eur Heart J. 2016. doi:10.1093/eurheartj/ehv689.
Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49(25):2379–93. doi:10.1016/j.jacc.2007.02.059.
Peng Y, Chen M, Liu XJ, Liu W, Li Q, Chai H, et al. The CYP2C19 genotype does not impact the long-term prognosis of patients with coronary artery disease. Atherosclerosis. 2013;227(1):106–11. doi:10.1016/j.atherosclerosis.2012.12.028.
Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. J Cell Sci. 2013;126(Pt 22):5101–9. doi:10.1242/jcs.138313.
Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. 2014;34(10):2191–8. doi:10.1161/ATVBAHA.114.303422.
Wang C, Baker BM, Chen CS, Schwartz MA. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol. 2013;33(9):2130–6. doi:10.1161/ATVBAHA.113.301826.
Chien S. Molecular and mechanical bases of focal lipid accumulation in arterial wall. Prog Biophys Mol Biol. 2003;83(2):131–51.
Himburg HA, Grzybowski DM, Hazel AL, LaMack JA, Li XM, Friedman MH. Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am J Physiol Heart Circ Physiol. 2004;286(5):H1916–22. doi:10.1152/ajpheart.00897.2003.
Koskinas KC, Chatzizisis YS, Papafaklis MI, Coskun AU, Baker AB, Jarolim P, et al. Synergistic effect of local endothelial shear stress and systemic hypercholesterolemia on coronary atherosclerotic plaque progression and composition in pigs. Int J Cardiol. 2013;169(6):394–401. doi:10.1016/j.ijcard.2013.10.021.
Panse N, Brett S, Panse P, Kareti K, Rewis D, Gilmore P, et al. Multiple plaque morphologies in a single coronary artery: insights from volumetric intravascular ultrasound. Catheter Cardiovasc Interv. 2004;61(3):376–80. doi:10.1002/ccd.10777.
Sasaki O, Nishioka T, Inoue Y, Isshiki A, Akima T, Toyama K, et al. Longitudinal heterogeneity of coronary artery distensibility in plaques related to acute coronary syndrome. Clin Res Cardiol. 2012;101(7):545–51. doi:10.1007/s00392-012-0424-6.
Choi G, Lee JM, Kim HJ, Park JB, Sankaran S, Otake H, et al. Coronary artery axial plaque stress and its relationship with lesion geometry: application of computational fluid dynamics to coronary CT angiography. J Am Coll Cardiol Img. 2015;8(10):1156–66. doi:10.1016/j.jcmg.2015.04.024.
Wald DS, Morris JK, Wald NJ, Chase AJ, Edwards RJ, Hughes LO, et al. Randomized trial of preventive angioplasty in myocardial infarction. N Engl J Med. 2013;369(12):1115–23. doi:10.1056/NEJMoa1305520.
Pollack A, Mohanty BD, Handa R, Looser PM, Fuster V, King III SB, et al. Preventive stenting in acute myocardial infarction. J Am Coll Cardiol Intv. 2015;8(1 Pt B):131–8. doi:10.1016/j.jcin.2014.09.006.
Dweck MR, Puntman V, Vesey AT, Fayad ZA, Nagel E. MR imaging of coronary arteries and plaques. J Am Coll Cardiol Img. 2016;9(3):306–16. doi:10.1016/j.jcmg.2015.12.003.
Pham TA, Hua N, Phinikaridou A, Killiany R, Hamilton J. Early in vivo discrimination of vulnerable atherosclerotic plaques that disrupt: a serial MRI study. Atherosclerosis. 2016;244:101–7. doi:10.1016/j.atherosclerosis.2015.11.013.
Phinikaridou A, Hua N, Pham T, Hamilton JA. Regions of low endothelial shear stress colocalize with positive vascular remodeling and atherosclerotic plaque disruption: an in vivo magnetic resonance imaging study. Circ Cardiovasc Imaging. 2013;6(2):302–10. doi:10.1161/CIRCIMAGING.112.000176.
Phinikaridou A, Ruberg FL, Hallock KJ, Qiao Y, Hua N, Viereck J, et al. In vivo detection of vulnerable atherosclerotic plaque by MRI in a rabbit model. Circ Cardiovasc Imaging. 2010;3(3):323–32. doi:10.1161/CIRCIMAGING.109.918524.
• Papafaklis MI, Mizuno S, Takahashi S, Coskun AU, Antoniadis AP, Tsuda M, et al. Incremental predictive value of combined endothelial shear stress, plaque necrotic core, and plaque burden for future cardiac events: a post-hoc analysis of the PREDICTION study. Int J Cardiol. 2016;202:64–6. doi:10.1016/j.ijcard.2015.08.208. The first study to report the significant additive predictive value of plaque tissue characteristics and local hemodynamic factors (low ESS) in indentifing plaques at high risk for adverse cardiovarcular events.
Stone P, Maehara A, Coskun AU, Maynard C, Andreou I, Siasos G et al. TCT-317 Local low endothelial shear stress (ESS) provides incremental prediction of non-culprit MACE in addition to plaque burden, minimal lumen area, and plaque morphology: the PROSPECT study. J Am Coll Cardiol. 2015;66(15_S). doi:10.1016/j.jacc.2015.08.333.
Zaromytidou M, Stone GW, Maehara A, Coskun AU, Maynard CC, Siasos G et al. Localization of Culprit Endothelial Shear Stress Patterns Along the Course of Untreated Coronary Lesions Responsible for Future Mace: The Prospect Study. Journal of the American College of Cardiology. 2016;67(13_S):358-. doi:10.1016/S0735-1097(16)30359-X.
Fukumoto Y, Hiro T, Fujii T, Hashimoto G, Fujimura T, Yamada J, et al. Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol. 2008;51(6):645–50. doi:10.1016/j.jacc.2007.10.030.
Gijsen FJ, Mastik F, Schaar JA, Schuurbiers JC, van der Giessen WJ, de Feyter PJ, et al. High shear stress induces a strain increase in human coronary plaques over a 6-month period. EuroIntervention. 2011;7(1):121–7. doi:10.4244/EIJV7I1A20.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marina Zaromytidou, Antonios P. Antoniadis, Gerasimos Siasos, Ahmet Umit Coskun, Ioannis Andreou, Michail I. Papafaklis, Michelle Lucier, Charles L. Feldman, and Peter H. Stone declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Marina Zaromytidou and Antonios P. Antoniadis contributed equally to this work and are joined first authors on this manuscript.
This article is part of the Topical Collection on Vascular Biology
Rights and permissions
About this article

Cite this article
Zaromytidou, M., Antoniadis, A.P., Siasos, G. et al. Heterogeneity of Coronary Plaque Morphology and Natural History: Current Understanding and Clinical Significance. Curr Atheroscler Rep 18, 80 (2016). https://doi.org/10.1007/s11883-016-0626-x
Published:
DOI: https://doi.org/10.1007/s11883-016-0626-x