
Abstract
Inflammation is a critical component in the development of coronary heart disease (CHD), specifically in the process of atherogenesis. Human translational and preclinical studies have demonstrated that inflammation contributes to the development, sustainment, and progression of atherosclerosis, and epidemiological studies demonstrate that human diseases associated with increased systemic inflammation increase the risk of CHD-related events. Therefore, over the last decade, multiple clinical studies were designed to target the inflammatory cascade in order to reduce the risk of CHD and to identify which populations may benefit from these preventative treatment strategies. This review briefly summarizes inflammation as a risk factor in atherosclerosis, human disease states associated with accelerated atherosclerosis, and current treatment strategies for CHD targeting the inflammatory cascade.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the 1880s, there was an emerging need to recognize and understand cardiovascular disease pathogenesis [1]. It was not until 1934, the International Society of Geographical Pathology held a conference describing data on the frequency of atherosclerotic lesions across countries ranked by occupation and social class [1]. From this, the first coronary heart disease (CHD) prospective study was initiated among a cohort of male professionals in Minnesota for a 15-year period, leading to the development of key investigations such as the Seven Countries Study and the Framingham Heart Study [2–4]. Although significant advances in the knowledge and understanding of CHD have emerged following these original studies, including identification of major CHD risk factors such as hypertension, hyperlipidemia, obesity, diabetes mellitus, and cigarette smoking, CHD remains the leading cause of death worldwide [5]. Additionally, up to half of all events associated with CHD are reported to occur in patients lacking traditional risk factors, resulting in the necessity to identify reliable biomarkers correlated with CHD risk and outcome, and shifting the perspective on the progression of atherogenesis [6, 7]. Atherosclerosis was initially described as a passive accumulation of lipids in the arterial wall, but is now acknowledged as a dynamic, immune-driven process culminating in the accumulation of oxidized lipids [8, 9]. The immunological processes found by immunohistochemistry and pathology supported that pro-inflammatory mechanisms are associated with the repair and healing process. In vitro cell culture, preclinical rodent models, and human translational and epidemiological studies have all contributed to the hypothesis that inflammation predisposes and drives CHD disease development in humans.
Inflammation, Cardiometabolic Diseases, and Atherosclerosis
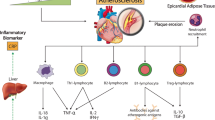
Atherosclerosis begins with some damage to the endothelial cell layer, which can be due to hypertension, diabetes, tobacco use, shear stress, or other emerging risk factors including chronic low-grade inflammation. The first response of endothelial cell activation is the expression of vascular cell adhesion molecules such as ICAM, VCAM, and E-selectin. The subsequent immune response begins with acute inflammatory cell infiltrates such as neutrophils, which heal the damaged endothelium and bind to the adhesion molecules. Following the cascade of neutrophil degranulation, chemotaxis of monocytes occurs to aid in this process. After a period of 3 to 7 days, chronic inflammatory cells including monocytes, macrophages, and T cells will arrive to the area to heal the injured wall. If the monocyte is retained in the intimal wall of the artery, it will become a resident tissue macrophage, which will scavenge lipids and glucose and promote further inflammatory cell infiltration. Soon, this macrophage with increasing lipid content will become a foam cell, which secretes pro-inflammatory cytokines including TNF-α, IFN-γ, and IL-6. A similar process occurs with T cells, which differentiate into Th1, IFN-γ-producing cells. Over the ensuing period of time, smooth muscle cells will proliferate and encroach on the lumen when the arterial wall cannot accommodate more inflammatory cells. This will soon change from a fatty streak to a luminal irregularity, which can be detected by imaging [9].
The term cardiometabolic disease encompasses most of the traditional risk factors of CHD (dyslipidemia, dysglycemia, hypertension), with each risk factor accompanied by an inflammatory component. For example, in an obese state, expressions of inflammatory cytokines such as TNFα, IL-6, IL-1β, and CCL2 are not limited to adipose tissue, but are also observed in the liver, pancreas, brain, and possibly muscle, leading to the development of metabolic syndrome [10–15]. Both epidemiological and human clinical studies suggest a causal link between chronic inflammation, insulin resistance, and type 2 diabetes [16, 17]. Compelling data using a low-dose human endotoxemia study demonstrated that the acute effects of innate immune-driven inflammation led to an insulin resistant, diabetic-like state shortly after injection of LPS in healthy humans [18]. Healthy men and women, receiving a low-dose of lipopolysaccharide, exhibited systemic insulin resistance 6 h post-injection, concomitant with an escalation of TNFα and IL-6 [19]. The observed systemic insulin resistance was independent of pancreatic β cell dysfunction [18] and suggested a temporal relationship with dose-response effects between inflammation and subsequent cardiometabolic diseases [18].
In September 1992, the recruitment for the Women’s Health Study, a primary prevention trial, began to determine if aspirin combined with vitamin E would reduce the incidence of myocardial infarction in a placebo-controlled study of 39,876 women over a period of 10 years [6]. The primary outcome included nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes [6]. Although it was determined, there was no significant decrease in the risk of major cardiovascular events in the aspirin group compared to the placebo group, and there was a 17 % reduction in the risk of stroke in the aspirin group. In addition, compared to placebo, aspirin had no effect on the risk of fatal or nonfatal infarction [6]. However, subgroup analysis revealed that in women over the age of 65, there was a decrease in the risk of major cardiovascular events, ischemic stroke, and myocardial infarction. Another significant outcome from the Women’s Health Study (WHS) was the observation that high-sensitivity (hs) C-reactive protein (CRP) level, a nonspecific protein marker of inflammation produced by the liver in response to innate immune activation, was a more accurate predictor of cardiovascular risk than LDL cholesterol, further supporting the hypothesis that CHD is an immune-driven inflammatory disease [20]. An association of hsCRP with risk for CVD has been described in many studies, the largest being the MRFIT (Multiple Risk Factor Intervention Trial). This showed a strong relationship between levels of CRP and mortality from CHD in high-risk middle-aged men [21]. In the context of other markers of inflammation and risk for CVD events, the Emerging Risk Factor Collaboration (ERFC) reviewed the association among hsCRP levels, CV risk factors, and vascular risks in 160,309 individuals from 54 prospective studies and found that CRP concentration was associated with increased risk of CHD, ischemic stroke, and death from vascular causes [22].
Human Inflammatory Diseases Associated with Atherosclerosis
Chronic inflammatory diseases exhibit elevated risk of early onset CHD beyond traditional risk factors. For example, rheumatoid arthritis is a chronic inflammatory disease of the joints that has an accelerated rate of atherogenesis compared to healthy patients [23, 24]. Multiple studies suggest that the degree and duration of inflammatory distress dictate early onset atherogenesis in rheumatoid arthritis. In one case-controlled study, rheumatoid arthritis patients had a 44 % increase in carotid atherosclerosis compared to non-rheumatoid arthritis patients, correlating specifically with disease duration and anti-TNF therapy [25]. In a meta-analysis pooled study, results from various parts of the world demonstrated a 48 % increase risk for cardiovascular disease in rheumatoid arthritis patients relative to the general population [26]. In addition, Giles et al. demonstrated that rheumatoid arthritis patients have an increased proportion of visceral fat, resulting in increased susceptibility to metabolic syndrome and elevated cardiovascular risk [27]. A nonspecific T cell inhibitor, methotrexate, reduces cardiovascular risk in rheumatoid arthritis patients, suggesting that chronic inflammation is primary key component in CHD [28, 29].
Patients suffering from systemic lupus erythematosus (SLE) display increased cardiovascular events with early onset, not explained by traditional risk factors such as aging, altered lipid levels, and smoking. In fact, there is a tenfold increase in the risk of SLE patients having a myocardial infarction compared to the general population [30]. Hydroxychloroquine, an SLE treatment, reduces plaque burden on carotid ultrasound images and seems to be thromboprotective with a 68 % reduction in the risk of thrombovascular events [31, 32]. Hydroxychloroquine disrupts the production of IFNα, an inflammatory cytokine involved in endothelial cell dysfunction, by interfering with TLR7 and TLR9 signaling [8, 33]. The ability of hydroxychloroquine to reduce vascular inflammation, hyperglycemia, and dyslipidemia provides atheroprotective effects. Rituximab, a biologic therapy that results in the depletion of B cells, exerts positive effects on the risk factors of atherogenesis in SLE. B cell depletion decreases inflammation, improves the lipid profile, and decreases disease activity, further suggesting a link between chronic inflammation in SLE and atherogenesis [34]. Taken together, targeting components of the inflammatory cascade may reduce CHD risk in SLE patients.
Psoriasis is a chronic inflammatory disorder of the skin affecting 2–4 % of the population and is associated with an increased risk of cardiovascular events, specifically atherogenesis [35–37]. The 58 % increased risk of major adverse cardiovascular events, myocardial infarction, stroke, and atherosclerosis, associated with psoriasis and psoriatic arthritis are a critical concern [37, 38]. Additionally, the psoriasis skin severity may be associated with systemic inflammation and the extent of cardiovascular disease [37, 38]. Mehta et al. and others recently demonstrated enhanced vascular inflammation in severe psoriasis patients compared to age-matched healthy controls, which was associated with increased neutrophil frequency and activation [39–41, 42•]. Observational studies demonstrate that the use of methotrexate and anti-TNF drugs reduces myocardial infarction [43–45]. Furthermore, anti-TNF drugs reduce vascular inflammation in psoriasis suggesting that a reduction of inflammation may impact vascular diseases and subsequent events [41].
HIV, a virus-induced chronic inflammatory disease state, exhibits increased cardiovascular risk also attributable to increased inflammation and immune activation [46, 47]. In HIV patients, endotoxemia is elevated and thought to be a potential mechanism of increased cardiovascular risk. The endotoxin lipopolysaccharide is a potent stimulator of monocytes and macrophages resulting in the activation and initiation of atherogenesis. Lipopolysaccharide is hypothesized to mediate chronic inflammation in HIV patients and serum CD14s level, a biomarker of monocyte activation through lipopolysaccharide interaction that correlates with atherosclerosis progression in HIV patients [48–50].
Treatment of Inflammation and Amelioration of CVD
The Justification for the Use of Statin in Prevention: An Intervention Trial Evaluating Rosuvastatin trial was designed to determine if lowering hsCRP would reduce the risk of cardiovascular events [51••, 52]. The JUPITER study, a primary prevention trial, was designed to assess whether high-dose statin therapy operating through anti-inflammatory pathways would reduce first cardiovascular event. The trial was a formal hypothesis testing trial based on previous observations that inflammation pays a critical role in atherogenesis, and hsCRP independently predicts vascular events independent of low-density lipoprotein cholesterol (LDL-c) levels, and that in acute coronary syndrome, the magnitude of the benefit of statins is associated with the levels of hsCRP [51••]. In order to test the hypothesis, 17,802 men and women with high levels of hsCRP (>2.0 mg/L) and low levels of LDL-c (<130 mg/dL) and triglycerides below 500 mg/dL were enrolled and given 20 mg per day of rosuvastatin. With a primary endpoint of myocardial infarction, stroke, arterial revascularization, hospitalization for unstable angina, or death from cardiovascular cause, JUPITER demonstrated that rosuvastatin reduced LDL-c by 50 % concomitant with a 37 % reduction in hsCRP. Overall, the trial was ended early due to a 44 % reduction in the primary end point of all vascular events, a 54 % reduction in myocardial infarction, a 48 % reduction in stroke, a 46 % reduction in need for arterial revascularization, and a 20 % reduction in all cause mortality, demonstrating a reduction of hsCRP, an inflammatory marker, positively correlates with CHD risk [51••]. Many have suggested that the intense lowering of events in the treatment arm was in part due to an intense lowering of LDL-c rather than a decrease in inflammation, which led to the development of two large ongoing studies to test the inflammatory hypothesis [53].
The Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) and Cardiovascular Inflammation Reduction Trial (CIRT) trials are designed to directly test the hypothesis that inflammation drives atherogenesis. In these studies, inflammatory components are directly targeted to determine if inhibition of inflammation will reduce cardiovascular event rates [54•]. CIRT utilizes a low-dose of methotrexate, a nonspecific T cell inhibitor, in a group of post-myocardial infarction patients with either diabetes or metabolic syndrome. This idea is based on the fact that psoriatic arthritis and rheumatoid arthritis patients on methotrexate have reduced cardiac events and decreased TNFα, IL-6, and CRP levels. The second trial, CANTOS, evaluates whether IL-1β inhibition with a human monoclonal antibody, compared to placebo, can reduce the rates of recurrent myocardial infarction, stroke, and cardiovascular death among stable coronary artery disease patients at high vascular risk due to hsCRP elevation. IL-1β is involved in inflammasome activation, and therefore, its blockade hypothetically would reduce the amount of inflammasome activation and subsequent immune responses observed in atherogenesis. The CANTOS and CIRT trials are designed to reduce inflammation independent of other concomitant pathways for vascular disease [54•].
Conclusions
In conclusion, the link between inflammation and the development of CHD is proving stronger with each study. Current and future treatment strategies targeting inflammatory components yield promising outcomes in the primary and secondary prevention of CHD; however, only rigorous, prospective ongoing trials will inform whether the relationship between inflammation and CHD is associative or causal.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat Rev Cardiol. 2014;11:276–89.
Keys A, Taylor HL, Blackburn H, Brozek J, Anderson JT, Simonson E. Coronary heart disease among Minnesota business and professional men followed fifteen years. Circulation. 1963;28:381–95.
Epstein FH. Cardiovascular disease epidemiology: a journey from the past into the future. Circulation. 1996;93:1755–64.
Wong ND, Levy D. Legacy of the Framingham Heart Study: rationale, design, initial findings, and implications. Glob Heart. 2013;8:3–9.
Torpy JM, Burke AE and Glass RM. Jama patient page. Coronary heart disease risk factors. JAMA. 2009;302:2388.
Ridker PM, Cook NR, Lee IM, Gordon D, Gaziano JM, Manson JE, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352:1293–304.
Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA. 2005;294:56–65.
Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE—mechanisms and management. Nat Rev Rheumatol. 2012;8:214–23.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49.
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–90.
Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–70.
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–9.
Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J Clin Invest. 1996;97:1111–6.
Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34:1809–15.
Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007;27:1433–9.
Mehta NN, Heffron SP, Patel PN, Ferguson J, Shah RD, Hinkle CC, et al. A human model of inflammatory cardio-metabolic dysfunction; a double blind placebo-controlled crossover trial. J Transl Med. 2012;10:124.
Anderson PD, Mehta NN, Wolfe ML, Hinkle CC, Pruscino L, Comiskey LL, et al. Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab. 2007;92:2272–9.
Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–65.
Kuller LH, Tracy RP, Shaten J and Meilahn EN. Relation of C-reactive protein and coronary heart disease in the MRFIT nested case-control study. Multiple risk factor intervention trial. Am J Epidemiol. 1996;144:537–47.
Emerging Risk Factors C, Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–40.
Nagata-Sakurai M, Inaba M, Goto H, Kumeda Y, Furumitsu Y, Inui K, et al. Inflammation and bone resorption as independent factors of accelerated arterial wall thickening in patients with rheumatoid arthritis. Arthritis Rheum. 2003;48:3061–7.
Giles JT, Post WS, Blumenthal RS, Polak J, Petri M, Gelber AC, et al. Longitudinal predictors of progression of carotid atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2011;63:3216–25.
Roman MJ, Moeller E, Davis A, Paget SA, Crow MK, Lockshin MD, et al. Preclinical carotid atherosclerosis in patients with rheumatoid arthritis. Ann Intern Med. 2006;144:249–56.
Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524–9.
Giles JT, Allison M, Blumenthal RS, Post W, Gelber AC, Petri M, et al. Abdominal adiposity in rheumatoid arthritis: association with cardiometabolic risk factors and disease characteristics. Arthritis Rheum. 2010;62:3173–82.
Krishnan E, Lingala VB, Singh G. Declines in mortality from acute myocardial infarction in successive incidence and birth cohorts of patients with rheumatoid arthritis. Circulation. 2004;110:1774–9.
Choi HK, Hernan MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359:1173–7.
Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du Berger R, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–7.
Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–406.
Jung H, Bobba R, Su J, Shariati-Sarabi Z, Gladman DD, Urowitz M, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 2010;62:863–8.
Sun S, Rao NL, Venable J, Thurmond R, Karlsson L. TLR7/9 antagonists as therapeutics for immune-mediated inflammatory disorders. Inflamm Allergy Drug Targets. 2007;6:223–35.
Pego-Reigosa JM, Lu TY, Fontanillo MF, del Campo-Perez V, Rahman A, Isenberg DA. Long-term improvement of lipid profile in patients with refractory systemic lupus erythematosus treated with B-cell depletion therapy: a retrospective observational study. Rheumatology (Oxford). 2010;49:691–6.
Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512–6.
Prodanovich S, Kirsner RS, Kravetz JD, Ma F, Martinez L, Federman DG. Association of psoriasis with coronary artery, cerebrovascular, and peripheral vascular diseases and mortality. Arch Dermatol. 2009;145:700–3.
Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31:1000–6.
Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735–41.
Mehta NN, Yu Y, Saboury B, Foroughi N, Krishnamoorthy P, Raper A, et al. Systemic and vascular inflammation in patients with moderate to severe psoriasis as measured by [18F]-fluorodeoxyglucose positron emission tomography-computed tomography (fdg-pet/ct): a pilot study. Arch Dermatol. 2011;147:1031–9.
Dave J, Ahlman MA, Lockshin BN, Bluemke DA, Mehta NN. Vascular inflammation in psoriasis localizes to the arterial wall using a novel imaging technique. J Am Acad Dermatol. 2014;70:1137–8.
Bissonnette R, Tardif JC, Harel F, Pressacco J, Bolduc C, Guertin MC. Effects of the tumor necrosis factor-alpha antagonist adalimumab on arterial inflammation assessed by positron emission tomography in patients with psoriasis: results of a randomized controlled trial. Circ Cardiovasc Imaging. 2013;6:83–90.
Naik HB, Natarajan B, Stansky E, Ahlman MA, Teague H, Salahuddin T, et al. Severity of psoriasis associates with aortic vascular inflammation detected by FDG PET/CT and neutrophil activation in a prospective observational study. Arterioscler Thromb Vasc Biol. 2015. doi:10.1161/ATVBAHA.115.306460. This manuscript is the first to show a direct relationship between the severity of skin disease and vascular disease by FDG PET/CT.
Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52:262–7.
Nguyen T, Wu JJ. Relationship between tumor necrosis factor-alpha inhibitors and cardiovascular disease in psoriasis: a review. Perm J. 2014;18:49–54.
Ahlehoff O, Skov L, Gislason G, Lindhardsen J, Kristensen SL, Iversen L, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273:197–204.
Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One. 2012;7, e44454.
Kelesidis T, Kendall MA, Yang OO, Hodis HN, Currier JS. Biomarkers of microbial translocation and macrophage activation: association with progression of subclinical atherosclerosis in HIV-1 infection. J Infect Dis. 2012;206:1558–67.
Morange PE, Tiret L, Saut N, Luc G, Arveiler D, Ferrieres J, et al. TLR4/Asp299Gly, CD14/C-260T, plasma levels of the soluble receptor CD14 and the risk of coronary heart disease: The PRIME Study. Eur J Hum Genet. 2004;12:1041–9.
Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199:1177–85.
Ratcliffe NR, Kennedy SM, Morganelli PM. Immunocytochemical detection of Fcgamma receptors in human atherosclerotic lesions. Immunol Lett. 2001;77:169–74.
Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto Jr AM, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. The JUPITER study is the first to show that anti-inflammatory effects by way of treatment with high dose statin may reduce first CV event.
Ridker PM. The JUPITER trial results, controversies, and implications for prevention. Circ Cardiovasc Qual Outcomes. 2009;2:279–85.
Shishehbor MH, Hazen SL. JUPITER to Earth: a statin helps people with normal LDL-c and high hs-CRP, but what does it mean? Cleve Clin J Med. 2009;76:37–44.
Ridker PM. Closing the loop on inflammation and atherothrombosis: why perform the CIRT and CANTOS trials? Trans Am Clin Climatol Assoc. 2013;124:174–90. The CIRT and CANTOS trials are the first studies designed to directly test whether reduction of inflammation with anti-inflammatory therapy reduces second myocardial infarction.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
H. Teague and N. N. Mehta declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Coronary Heart Disease
Rights and permissions
About this article

Cite this article
Teague, H., Mehta, N.N. The Link Between Inflammatory Disorders and Coronary Heart Disease: a Look at Recent Studies and Novel Drugs in Development. Curr Atheroscler Rep 18, 3 (2016). https://doi.org/10.1007/s11883-015-0557-y
Published:
DOI: https://doi.org/10.1007/s11883-015-0557-y