
Abstract
Robust epidemiologic and genetic studies have solidified the role of lipoprotein (a) [Lp(a)] as an independent and causal risk factor for cardiovascular disease. The increased cardiovascular risk of Lp(a) is mediated through both proatherogenic and prothrombotic/antifibrinolytic mechanisms. Several societies recommend Lp(a) screening for patients with high cardiovascular risk, although no consensus exists on the management of patients with elevated Lp(a). However, numerous pharmacologic approaches are being evaluated that have the potential to reduce Lp(a) and will be the focus of this review. The majority of these interventions have been developed for other lipid-lowering indications, but also lower Lp(a). There are also novel therapies in development that specifically target Lp(a). The efficacy of these therapies varies, and their role in the evolving lipoprotein therapeutic landscape has yet to be determined. Nevertheless, targeted Lp(a) reduction is certainly intriguing and will likely continue to be an active area of investigation in the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is a major cause of morbidity and mortality in the USA. Screening, diagnosis, and management of dyslipidemias play an important role in modifying CVD risk. Cholesterol screening in the general population is often limited to measurement of total cholesterol, high-density lipoprotein cholesterol (HDL-C), triglycerides, and either calculation or direct measurement of low-density lipoprotein cholesterol (LDL-C). However, lipoprotein (a) [Lp(a)] is an atherogenic lipoprotein particle that deserves attention, particularly in patients with the highest cardiovascular risk [1••]. Recently, Lp(a) has reemerged as a particle of particular clinical interest due to robust epidemiologic and human genetic studies that have solidified its role as an independent and causal risk factor for CVD, including myocardial infarction (MI), coronary death, ischemic stroke, and aortic valve calcification [2–6].
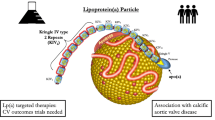
The increased cardiovascular risk of Lp(a) is mediated through both proatherogenic and prothrombotic/antifibrinolytic mechanisms. Lp(a) structurally resembles low-density lipoprotein cholesterol and consists of an apolipoprotein (a) [apo(a)] molecule covalently bound to apolipoprotein B-100 (apoB) (Fig. 1). Apo(a) has structural homology with plasminogen but has no fibrinolytic activity. Lp(a) is thought to be atherogenic starting at levels greater than 25–30 mg/dl, although this association is strongest at the highest levels of Lp(a), especially above 50 mg/dl [7]. In the Copenhagen City Heart Study, patients in the general population with the highest levels of Lp(a) (≥120 mg/dl) had more than a threefold risk of developing MI compared to those with the lowest Lp(a) levels [5]. High Lp(a) levels are also associated with increased incidence of ischemic stroke in blacks and white women [8]. Importantly, elevated Lp(a) is also an independent predictor of CVD risk in patients with familial hypercholesterolemia, independent of LDL-C levels [9].

Schematic drawing of the structure of Lp(a). Lp(a) consists of an apo(a) molecule covalently bound to apoB by a disulfide bond. Heterogeneity in the number of kringle IV (KIV) type 2 repeats accounts for different apo(a) isoforms. The homology shared by apo(a) and plasminogen is one postulated mechanism explaining the prothrombotic effects of Lp(a). Reprinted from [59], with permission from Elsevier
Atherogenicity of Lipoprotein (a)
The exact biologic role of the Lp(a) particle is not fully elucidated. However, it is thought to mediate CVD risk through both proatherogenic and prothrombotic effects. From a proatherogenic standpoint, not only does Lp(a) structurally resemble LDL particles (Fig. 1) but it may also be more avidly retained within the arterial wall than LDL-C [10]. Lp(a) has also been shown to be a preferential carrier of proinflammatory oxidized phospholipids (OxPL), which accumulate in atherosclerotic lesions and mediate plaque destabilization [11–13]. OxPL/apoB levels, in turn, are independently correlated with angiographically documented coronary artery disease and carotid and femoral atherosclerosis [11, 14]. The correlation of OxPL/apoB to Lp(a) is strongest in patients with the smallest apo(a) isoforms [15]. This strong association might explain, in part, why the smaller isoforms are more atherogenic [13, 15]. Other mechanisms by which Lp(a) may promote atherogenesis are by increasing endothelial cell permeability, promoting smooth muscle proliferation, and macrophage foam cell formation [11, 16].
Lp(a) is also postulated to promote thrombosis through various mechanisms. Apo(a) has been found to be strikingly homologous to plasminogen, suggesting that apo(a) may be able to interfere with the conversion of plasminogen to plasmin, thereby inhibiting fibrin clot lysis [16]. Smaller Lp(a) isoforms also bind more avidly to fibrin, thereby inhibiting plasmin formation to a greater extent [17]. Lp(a) can also bind to, and inactivate, tissue factor pathway inhibitor, which is a major regulator of the tissue-factor mediated coagulation pathway [16]. This combination of proatherogenic and prothrombotic properties of Lp(a) accounts for the unique and high-risk nature of this lipoprotein particle.
Determinants of Lipoprotein (a) Levels
Lp(a) levels are primarily genetically determined [1••]. Two independent single-nucleotide polymorphisms (SNPs) in the apo(a) gene LPA, rs3798220 and rs10455872, explain 36 % of the variation in Lp(a) levels [18]. These two SNPs are associated with elevated Lp(a) levels and an increased risk of coronary heart disease (CHD) [19]. In fact, women who were carriers of the rs3798220 allele had a twofold higher risk of major cardiovascular events compared to non-carriers in the Women’s Health Study [20]. A third SNP, rs9457951, is associated with increased Lp(a) levels in African-Americans and explains about 5 % of Lp(a) variance in African-Americans [21]. Of note, Lp(a) levels have high ethnic variability. In the Atherosclerosis Risk in Communities (ARIC) study, a robust epidemiological study, median Lp(a) levels were almost three times higher in blacks compared to whites (12.8 mg/dl compared with 4.3 mg/dl) [22]. In addition, Lp(a) levels >25 mg/dl are found in 60 to 70 % of African-Americans compared with 30 % of Caucasians [1••, 23].
Lp(a) size, like serum level, is also genetically determined and is related to the atherogenicity of the particle. Lp(a) formation occurs on the surface of hepatocytes, where an apo(a) “kringle” moiety forms a disulfide bond with apoB [24, 25] (Fig. 1). Variability in the number of kringle IV type 2 repeats in the LPA gene that encodes apo(a) explains the size variation of apo(a), which ranges from 187 to greater than 662 kDa [26]. Heterogeneity in apo(a) size is important, as patients with smaller apo(a) isoforms have a twofold higher risk of CHD compared to those with larger isoforms [27].
Measurement of Lipoprotein (a)
Currently, there are several commercial assays available to measure Lp(a), including immunoturbidimetry, nephelometry, and enzyme-linked immunosorbent assays. However, the structural complexity and size heterogeneity of Lp(a) have hindered the development of assays that are able to accurately measure Lp(a) concentrations in plasma [28]. The ideal assay would be apo(a) “isoform-insensitive,” although at this time there are no commercially available assays that are completely insensitive to the variability in apo(a) isoform mass as well as the lipid mass [29•]. The International Federation of Clinical Chemistry has developed a reference material for Lp(a), which measures Lp(a) in nanomoles per liter [28]. Other assays also measure Lp(a) as “mass” in milligrams per deciliter, which represents the entire mass of Lp(a), including lipids, proteins, and carbohydrates, while others measure Lp(a) as “cholesterol” in milligrams per deciliter [1••, 29•]. At this time, societies that make recommendations for Lp(a) screening generally report Lp(a) levels in milligrams per deciliter and generally use an upper limit of normal of 30 mg/dl [30, 31]. Assays that report Lp(a) in nanomoles per liter generally use an upper limit of normal of 75 nmol/l.
In addition to the use of a validated and standardized assay, racial- and ethnic-based reference ranges will be imperative if Lp(a) measurement and target therapies are to become more widespread. Studies that have examined Lp(a) levels in non-white/non-black populations have not uniformly shown an independent association with increased CVD risk [32–34]. Population and epidemiologic studies looking at varied racial and ethnic groups are greatly needed at this time to help determine population ranges of Lp(a) levels and association with increased CVD risk.
Screening for Elevated Lipoprotein (a)
Recommendations for Lp(a) screening are also not standardized among lipid, atherosclerosis, and cardiovascular preventive societies. The 2013 American College of Cardiology/American Heart Association cholesterol treatment guideline did not examine the role of Lp(a) and therefore gives no recommendations either for or against Lp(a) screening [35]. In contrast, both the National Lipid Association (NLA) and European Atherosclerosis Society (EAS) Consensus Panel suggest that the following groups of patients be screened for elevated Lp(a) levels: those with a personal or family history of premature CVD, familial hypercholesterolemia (FH), recurrent CVD events despite statin therapy, ≥3 % 10-year risk of fatal CVD according to European guidelines, and ≥10 % 10-year risk of fatal and/or non-fatal CHD according to US guidelines [30, 31]. It should be noted that the NLA does not recommend checking Lp(a) levels in low-risk patients (<5 % 10-year Framingham risk) [30]. The 2012 Canadian Cardiovascular Society recommends consideration of secondary testing, including Lp(a) measurement, in patients with a 10–19 % Framingham Risk Score but emphasizes that LDL-C reduction is the primary target in dyslipidemia management [36].
Addressing Cardiovascular Risk in Patients with Elevated Lp(a)
Studies suggest that the risk of CVD associated with elevated Lp(a) levels is even higher in the presence of other risk factors, including elevated LDL-C [37]. In a post hoc analysis of the Familial Atherosclerosis Treatment Study, in those patients with very elevated LDL-C levels, the best correlate of baseline CAD severity was elevated Lp(a). In patients who achieved a “substantial” LDL-C reduction (mean reduction of 40 %), persistent elevations in Lp(a) were no longer clinically significant in terms on CVD events [38]. Based on this and other robust data showing CVD event reduction with substantial LDL-C lowering, statins should be considered first-line therapy for patients with elevated LDL-C and elevated Lp(a). Current guidelines recommend that statins should be used as a first-line lipid-lowering therapy for cardiovascular risk reduction in patients with clinical atherosclerotic cardiovascular disease (ASCVD), diabetics aged 40–74, those with elevated cardiovascular risk using the 10-year Pooled Cohorts Equations, and those with primary severe elevations in LDL-C (>190 mg/dl) [35]. Specifically in patients with very high LDL-C levels suggesting familial hypercholesterolemia, Lp(a) is an additive risk factor [9, 39•]. Therefore, most patients who qualify for Lp(a) screening and are found to have elevated Lp(a) will also have indications for statin therapy, and the goal should be to aggressively lower LDL-C [39•]. However, it should be noted that there is an ongoing controversy about specific cholesterol targets as well as conflicting guidelines. In patients who have primary elevations in Lp(a) without an indication for LDL-C therapy, the benefit of statin therapy for risk reduction is unclear.
In addition to statin therapy, aspirin therapy should be considered in patients with Lp(a) to lower the overall cardiovascular risk. Historically, there was one small study showing a robust reduction in Lp(a) levels with aspirin, although no subsequent studies have been able to reproduce those results [40]. With regard to CVD risk reduction, post hoc analysis of the Women’s Health Study found that women (mean age 52) who were carriers of the higher-risk rs3798220 allele who were taking alternate-day 100 mg aspirin had a 56 % risk reduction of major CV events including myocardial infarction, ischemic stroke, or CV death (age-adjusted HR = 0.44, 95 % CI 0.20–0.94) [20]. In comparison, non-carriers who also received aspirin with the same dosing schedule had a non-significant risk reduction [20]. However, this study was neither powered to determine whether differences in Lp(a) level explained the CV events among rs3798220 carriers nor did it examine whether aspirin reduced Lp(a) levels. Based on current preventive guidelines, many high-risk patients with elevated Lp(a) may qualify for aspirin therapy for cardiovascular event reduction, independent of any effects on Lp(a).
Therapies to Lower Lipoprotein (a)
While it is currently unknown whether lowering plasma Lp(a) will result in decreased cardiovascular mortality, the significant CVD risk associated with increased Lp(a) levels has led to investigation into pharmacologic approaches to lower Lp(a) levels. We review pharmacologic approaches, both approved and in development, that have the potential to reduce Lp(a) levels. The majority of these interventions have been developed for other lipid lowering indications, but also lower Lp(a). There are also novel therapies in development to specifically target Lp(a).
An approved pharmacologic agent that is often recommended for patients with elevated Lp(a) is nicotinic acid. In addition, apheresis is not only approved for use in patients with very elevated LDL-C but also does lower Lp(a). Estrogen lowers Lp(a) but is not recommended for CVD prevention. Although approved only for homozygous familial hypercholesterolemia, lomitapide and mipomersen also lower Lp(a), likely by impairing atherogenic lipoprotein production. Novel therapies in development for other lipid-lowering indications that also show promise for reducing Lp(a) include proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitorsand cholesteryl ester transfer protein (CETP) inhibitors. Additional therapies that are in early development which demonstrate the potential for Lp(a)-lowering include thyrominetics, farnesoid X receptor (FXR) agonists, and antibodies to interleukin-6 (IL-6). Finally, there is an antisense oligonucleotides to apo(a) in development which directly targets Lp(a). These approaches are summarized in Tables 1 and 2 and will be further described in the sections below.
Available Therapies
Nicotinic Acid
Nicotinic acid (niacin, vitamin B3) is a water-soluble vitamin that has been available for almost 60 years for the treatment of dyslipidemia and which has been shown in the pre-statin era to significantly decrease the incidence of both MI and all-cause mortality [41–43]. At therapeutic doses, nicotinic acid lowers LDL-C, raises HDL-C, lowers triglycerides, and also lowers Lp(a). With regard to Lp(a) specifically, older studies have demonstrated as much as 30–40 % reduction in levels with nicotinic acid [44, 45]. However, given the multifaceted lipid effects of nicotinic acid, no study has yet to definitively correlate Lp(a) reductions from nicotinic acid with improved CVD outcomes.
Post hoc analysis of the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes) study, a prospective, randomized double-blind trial of 3414 patients at high CVD risk on background statin therapy with low baseline LDL-C levels, found a modest 19 % Lp(a) reduction in those treated with extended-release nicotinic acid compared to placebo (median baseline Lp(a) 32.6 nmol/l in the placebo group, 36.0 nmol/l in the nicotinic acid group) [46]. However, no reduction in major CVD event rates was demonstrated despite favorable reductions in Lp(a) [46]. The subsequent, and much larger, HPS-2-THRIVE (Heart Protection Study-2—Treatment of HDL to Reduce the Incidence of Vascular Events) study of 25,673 adults with vascular disease also on background statin therapy with low baseline LDL-C levels found a significant decrease in Lp(a) levels at 1 year in a small subset of 1999 patients (50.7 nmol/l in the niacin/lapropriprant group versus 60.3 nmol/l in the placebo group—no baseline values given) [47]. However, HPS-2-THRIVE also failed to show a significant benefit in reducing major vascular events with nicotinic acid compared to placebo.
To date, we are still unsure of the role of nicotinic acid for CVD event reduction when the LDL-C level is already at low levels on statin therapy. However, a recent meta-analysis suggests that nicotinic acid is still a useful therapy for CVD event reduction [48]. For patients with elevated Lp(a) specifically, based on the available evidence, the EAS recommends the use of nicotinic acid at a dose of 1–3 g/day in the highest risk patients to achieve Lp(a) levels less than 50 mg/dl as a secondary priority after reductions in LDL-C and total cholesterol levels have been achieved [31].
Apheresis
Apheresis is currently the most effective treatment modality to lower Lp(a) [49]. At this time, most lipid societies recommend apheresis only for severe hypercholesterolemia usually seen in patients with familial hypercholesterolemia [50, 51]. However, the German Committee of Physicians and Health Insurance Funds and England’s HEART-UK also recommend apheresis for an Lp(a) level greater than 60 mg/dl with progressive CHD [52, 53]. Jaeger et al. conducted the first longitudinal cohort study of 120 patient that investigated whether lipid apheresis along with lipid-lowering medication could reduce extremely high levels of Lp(a) more efficaciously than lipid-lowering medications alone [54]. Median Lp(a) concentration was reduced by 73 % with apheresis treatment with a corresponding significant 86 % reduction in major adverse coronary events compared with lipid-lowering therapy alone. A subsequent prospective, observational study of 170 patients with Lp(a)-hyperlipoproteinemia found that a single apheresis treatment reduced Lp(a) levels by up to 70 % and reduced the mean annual rate of major cardiovascular events by 78 % [52]. Importantly, given that apheresis removes Lp(a) and LDL-C particles with similar efficacy, the therapeutic effects seen in these studies may be related to reductions in Lp(a), LDL-C, or both lipoproteins. A notable limitation of apheresis is that both Lp(a) and LDL-C levels generally rebound to baseline levels within 2 weeks of treatment [52, 54]. Lp(a) levels, in particular, can rebound to 80 % of baseline before the subsequent apheresis session [52]. Although effective, apheresis has significant limitations including cost, limited access to centers, and the time commitment required for biweekly sessions of 2 to 4 h each [50].
Estrogens
In the 1990s, studies found that hormone replacement therapy (HRT) in postmenopausal women may lead to Lp(a) level reductions of up to 50 % [55–58]. However, most of these studies were conducted in small cohorts of women with a relatively short follow-up period. Mechanistically, it is possible that HRT accelerates Lp(a) catabolism by increasing LDL-C receptor-mediated clearance [57, 58]. However, kinetics studies suggest that Lp(a) clearance is not entirely dependent on the LDL-C receptor [59]. Since the results of the Women’s Health Initiative and the HERS I and HERS II studies demonstrated that HRT does not uniformly confer cardiac protection and may even increase early risk of CHD, the use of estrogen is currently not recommended for prevention of CVD [60–63].
Therapies Approved for Homozygous Familial Hypercholesterolemia
Mipomersen
Mipomersen (Kynamro, Genzyme, Cambridge, MA) is an apoB antisense oligonucleotide (ASO) that is FDA approved for lowering LDL-C, total cholesterol, apoB, and non-HDL-C in patients with homozygous familial hypercholesterolemia [64]. ASOs are short, single-stranded synthetic analogs of natural nucleic acids designed to bind to a specific target messenger RNA and suppress synthesis of a target protein [65•]. Mipomersen targets apoB and impairs the formation of very-low-density lipoproteins within hepatocytes and lowers LDL-C by ~25 % [64]. Specifically for Lp(a), a recent meta-analysis examining 444 patients from six phase 2 or phase 3 randomized controlled trials found a 26 % reduction in Lp(a) in patients receiving subcutaneous mipomersen 200 mg weekly compared with placebo [66]. Due to concerns for hepatoxicity, mipomersen can only be prescribed by certified providers. It is unclear if or how the LDL-C- versus Lp(a)-lowering effects of this compound will translate into reduction in clinical events in this very high-risk homozygous familial hypercholesterolemia population.
Lomitapide
Lomitapide (Juxtapid, Aegerion, Cambridge, MA) is an inhibitor of microsomal triglyceride transfer protein (MTP) that is FDA approved as an adjunct to diet and lipid-lowering therapies to lower LDL-C, total cholesterol, apoB, and non-HDL in patients with homozygous familial hypercholesterolemia [67]. MTP is an intracellular protein found in the endoplasmic reticulum of hepatocytes and enterocytes that is responsible for transferring lipid molecules onto apoB [68]. Inhibition of MTP impairs the formation of VLDL particles in hepatocytes and chylomicron particles in enterocytes and lowers LDL-C by ~40 % [69].
A phase 2, prospective, randomized, double-blind study of 84 hypercholesterolemic patients showed an Lp(a) reduction of 17 % in those receiving lomitapide (5 mg daily, titrated to 10 mg over the 12-week study) [68]. A subsequent single-arm, open-label phase 3 study looking at the efficacy and safety of lomitapide in patients with homozygous FH showed a 15 % reduction in Lp(a) levels at 26 weeks that persisted to 56 weeks (median dose of 40 mg daily) [70]. By week 78, however, there was no significant Lp(a) difference from baseline. The reason behind this loss of significance is unclear. As with mipomersen, due to concerns for hepatoxicity, lomitapide can only be prescribed by certified providers, and the long-term role of this therapy for Lp(a) reduction remains to be seen.
Therapies in Development with Potential Lp(a)-Lowering Effects
Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors
PCSK9, the most recently discovered member of the proprotein convertase family, is expressed in different cell types, but its expression in hepatocytes has created the most interest from a pharmacological prospective [71•]. PCSK9 mediates LDL receptor degradation via both intra- and extracellular pathways [71•]. Loss-of-function mutations and missense mutations in the PCSK9 gene have been linked to low plasma LDL-C levels and increased statin response, whereas gain-of-function mutations lead to an FH phenotype with very elevated LDL-C [72]. Inhibition of PCSK9 synthesis or function therefore represents an attractive therapeutic strategy to effectively decrease LDL-C levels. Currently, the use of monoclonal antibodies is the most promising pharmacologic method of inhibiting PCSK9-mediated degradation of LDL-C receptors and has demonstrated substantial LDL-C reductions [73].
In addition to lowering LDL-C, PCSK9 inhibitors have also been shown to lower Lp(a) levels. The mechanism by which PCSK9 inhibitors lower Lp(a) is currently under investigation, although it is postulated to be through decreased Lp(a) production or increased clearance [73]. The PROFICIO (Program to Reduce LDL and CV Outcomes Following Inhibition of PCSK9 In Different Populations) analyzed 1359 patients from four randomized, double-blind phase 2 trials looking at the effects of evolocumab (Amgen, Thousand Oaks, CA), a monoclonal antibody to PCSK9, compared to placebo, ezetimibe alone, or evolocumab with ezetimibe over a 12-week period [74]. In this pooled analysis with median baseline Lp(a) levels of 40.0 nmol/l, subcutaneous evolocumab treatment resulted in highly significant dose-related reductions in Lp(a) of up to 29.5 % with no plateau effect. A second monoclonal antibody, alirocumab (Regeneron, Tarrytown, NY), which is also administered subcutaneously, showed similar results to evolocumab in reducing Lp(a) levels. Pooled data from three double-blind, randomized, placebo-controlled phase 2 studies of 352 patients with hypercholesterolemia on background lipid-lowering therapy (statin or statin plus ezetimibe) showed a 30 % reduction in Lp(a) in the alirocumab group compared to 0.3 % in the placebo group (median baseline Lp(a) 19 mg/dl in the placebo group and 30 mg/dl in the alirocumab group) [75].
It remains to be seen where PCSK9 inhibitors will fit into the current therapeutic landscape and if this therapy will be used to specifically lower Lp(a) in addition to the robust LDL-C reductions that have been demonstrated.
Cholesteryl Ester Transfer Protein Inhibitors
CETP is a plasma protein that transfers cholesteryl esters from HDL-C to apoB-containing lipoproteins. CETP inhibitors significantly raise HDL-C levels, although the development of the first CETP inhibitor, torcetrapib, was prematurely terminated after the ILLUMINATE (Investigation of Lipid Management to Understand its Impact in Atherosclerotic Events) trial found that torcetrapib caused an excess of deaths and CV events, possibly related to off-target effects that increased aldosterone levels and blood pressure [76]. The development of a second CETP inhibitor, dalcetrapib, was also terminated after the phase 3 dal-OUTCOMES study in patients with recent acute coronary syndrome failed to demonstrate a significant reduction in recurrent CV events [77].
Two other CETP inhibitors, anacetrapib and evacetrapib, have shown beneficial lipid effects without the adverse off-target effects of torcetrapib. In the DEFINE (Determining the Efficacy and Tolerability of CETP Inhibition with Anacetrapib) trial, a phase 3 efficacy and safety study, anacetrapib raised HDL-C by 138 % and also lowered LDL-C by 40 % compared with placebo. In addition, anacetrapib was shown to lower Lp(a) levels by 36 % at 46 weeks, with a sustained reduction of 39 % at 76 weeks (median baseline Lp(a) 25.9 nmol/l in the placebo group, 26.8 nmol/l in the anacetrapib group) [78]. The mechanism by which CETP inhibition lowers Lp(a) is unknown. The REVEAL (Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification) trial is a phase 3 study involving 30,000 patients designed to evaluate whether anacetrapib will reduce major CVD events in patients on background statin therapy [79]. Completion of this study is expected by 2017. Evacetrapib has also been found to raise HDL-C by 45–129 % in a dose-dependent fashion and lower LDL-C by 14–36 % compared to placebo [80]. The ACCELERATE (Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition with Evacetrapib in Patients at a High-Risk for Vascular Outcomes) trial is a phase 3 study involving 12,000 patients designed to evaluate whether evacetrapib will reduce the first major CV event in patients with known vascular disease. Completion of this study is expected by 2016 [81]. As with all therapies in development, it remains to be seen if and where CETP inhibitors will fit into the current therapeutic landscape.
Thyromimetics
Thyroid hormones can stimulate cholesterol and lipoprotein metabolism via the thyroid receptor TRβ, which is predominantly expressed in the liver, and may also promote reverse cholesterol transport [82]. Thyroid hormone analogs, also known as thyromimetics, were developed to exert favorable lipid-lowering effects without disrupting the normal hypothalamic-pituitary-thyroid axis. One of these agents, eprotirome, initially held promise in decreasing Lp(a) in a dose-dependent fashion by up to 43 % in a phase 2 trial without adverse effects on the heart, bone, or pituitary (median baseline Lp(a) 36 mg/dl in the placebo group and 27–36 mg/dl in the eprotirome group) [83]. However, a subsequent phase 3 trial was prematurely terminated due to an animal study showing that eprotirome caused cartilage damage in dogs, and to the best of our knowledge, there are no further studies in process. Sobetirome, another thyromimetic, has also been reported to lower LDL-C without adverse effects, but has not been evaluated in clinical trials to date [82].
Farnesoid X Receptor
A preclinical pharmacologic target is FXR, which is a bile-activated transcription factor that has been shown to suppress apo(a) gene expression in mice and humans who have high bile acid levels [84, 85]. However, these early studies did not examine whether an agonist of FXR would directly reduce Lp(a) levels.
IL-6 Receptor Antagonists
Finally, there is a suggestion that Lp(a) plays a role in the acute phase response, a process in which IL-6 plays a major role [86]. Interestingly, a SNP (174G/C) in the IL-6 gene is associated with elevated Lp(a) levels [87]. Lp(a) levels were reduced by 30 % in a study of rheumatoid arthritis patients treated with tocilizumab, a monoclonal antibody against the IL-6 receptor [88]. It should be noted, however, that this was a study of just 11 patients. Nevertheless, the idea of Lp(a) as an acute phase reactant is an intriguing concept requiring further investigation.
Targeted Lp(a) Therapy in Early Development
In order to fully elucidate the role of Lp(a) modulation on CVD outcomes, a therapy that is specific for Lp(a) is required, as all of the other therapies discussed so far also modulate other lipoproteins. The first such therapy to our knowledge is an ASO directly targeting apo(a), akin to mipomersen targeting apoB. In pre-clinical mouse models, ASO 144367 led to significant reductions in both apo(a) and Lp(a) as well as their associated OxPL [89]. Lp(a) levels were reduced by 24.8 % and apo(a) levels by 19–93 %, with a more potent effect seen in mice expressing apo(a) with multiple KIV-2 repeats [89]. Evaluation of such therapies and evaluation of clinical outcomes with such therapies will be critical in fully establishing Lp(a) as a direct therapeutic target.
Conclusions
Lp(a) has reemerged as a particle of particular clinical interest due to robust epidemiologic and human genetic studies that have solidified its role as an independent and causal risk factor for CVD, including MI, coronary death, and ischemic stroke. With the complexities of the Lp(a) particle clearly defined, as well as new genetic-based therapeutic strategies in our armamentarium, we are now poised to start the journey towards fundamental breakthroughs in Lp(a) therapeutics. Currently, there is no therapy available that selectively decreases Lp(a) levels without affecting other lipoproteins, so it is still unknown whether lowering Lp(a) levels independent of LDL-C lowering will have a meaningful clinical effect. Therapies such as ASOs to apo(a) hold great hope and promise. Continued characterization of the Lp(a) particle will almost certainly bring further advances in diagnostic testing for screening purposes as well as therapeutics, and we will then need clinical outcome data to help determine whether these interventions provide a significant or incremental benefit. For now, the optimal treatment for patients with elevated Lp(a) is unknown, although aggressive risk factor modification with proven therapies is warranted for all high-risk patients with elevated Lp(a).
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60(8):716–21. This is a comprehensive and up to date review that highlights the role of Lp(a) in CVD risk and introduces recent therapies to lower Lp(a).
Bennet A, Di Angelantonio E, Erqou S, et al. Lipoprotein(a) levels and risk of future coronary heart disease: large-scale prospective data. Arch Intern Med. 2008;168(6):598–608.
Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–23.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–9.
Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176–84.
Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–12.
Kolski B, Tsimikas S. Emerging therapeutic agents to lower lipoprotein (a) levels. Curr Opin Lipidol. 2012;23(6):560–8.
Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407–12.
Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63(19):1982–9.
Nielsen LB. Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis. 1999;143(2):229–43.
Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5(5):673–94.
Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353(1):46–57.
Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49(10):2230–9.
Tsimikas S, Kiechl S, Willeit J, et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the Bruneck study. J Am Coll Cardiol. 2006;47(11):2219–28.
Tsimikas S, Witztum JL. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr Opin Lipidol. 2008;19(4):369–77.
Marcovina SM, Koschinsky ML. Evaluation of lipoprotein(a) as a prothrombotic factor: progress from bench to bedside. Curr Opin Lipidol. 2003;14(4):361–6.
Kang C, Dominguez M, Loyau S, Miyata T, Durlach V, Angles-Cano E. Lp(a) particles mold fibrin-binding properties of apo(a) in size-dependent manner: a study with different-length recombinant apo(a), native Lp(a), and monoclonal antibody. Arterioscler Thromb Vasc Biol. 2002;22(7):1232–8.
Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28.
Li Y, Luke MM, Shiffman D, Devlin JJ. Genetic variants in the apolipoprotein(a) gene and coronary heart disease. Circ Cardiovasc Genet. 2011;4(5):565–73.
Chasman DI, Shiffman D, Zee RY, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203(2):371–6.
Deo RC, Wilson JG, Xing C, et al. Single-nucleotide polymorphisms in LPA explain most of the ancestry-specific variation in Lp(a) levels in African Americans. PLoS One. 2011;6(1):e14581.
Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241–9.
Matthews KA, Sowers MF, Derby CA, et al. Ethnic differences in cardiovascular risk factor burden among middle-aged women: Study of Women’s Health Across the Nation (SWAN). Am Heart J. 2005;149(6):1066–73.
Kathiresan S. Lp(a) lipoprotein redux—from curious molecule to causal risk factor. N Engl J Med. 2009;361(26):2573–4.
Utermann G. The mysteries of lipoprotein(a). Science. 1989;246(4932):904–10.
Marcovina SM, Albers JJ, Scanu AM, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000;46(12):1956–67.
Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55(19):2160–7.
Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: recent advances and future directions. Clin Chem. 2003;49(11):1785–96.
McConnell JP, Guadagno PA, Dayspring TD, Hoefner DM, Thiselton DL, Warnick GR, et al. Lipoprotein(a) mass: a massively misunderstood metric. J Clin Lipidol. 2014;8:550–3. This brief review highlights the difficulty of Lp(a) measurement given the complexity of the molecule.
Davidson MH, Ballantyne CM, Jacobson TA, et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. J Clin Lipidol. 2011;5(5):338–67.
Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–53.
Wang W, Hu D, Lee ET, et al. Lipoprotein(a) in American Indians is low and not independently associated with cardiovascular disease. The Strong Heart Study. Ann Epidemiol. 2002;12(2):107–14.
Banerjee D, Wong EC, Shin J, Fortmann SP, Palaniappan L. Racial and ethnic variation in lipoprotein (a) levels among Asian Indian and Chinese patients. J Lipids. 2011;2011:291954.
Tavridou A, Unwin N, Bhopal R, Laker MF. Predictors of lipoprotein(a) levels in a European and South Asian population in the Newcastle Heart Project. Eur J Clin Invest. 2003;33(8):686–92.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–45.
Anderson TJ, Gregoire J, Hegele RA, et al. 2012 update of the Canadian Cardiovascular Society guidelines for the diagnosis and treatment of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2013;29(2):151–67.
Luc G, Bard JM, Arveiler D, et al. Lipoprotein (a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis. 2002;163(2):377–84.
Maher VM, Brown BG, Marcovina SM, Hillger LA, Zhao XQ, Albers JJ. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA. 1995;274(22):1771–4.
Jacobson TA. Lipoprotein(a), cardiovascular disease, and contemporary management. Mayo Clin Proc. 2013;88(11):1294–311. Comprehensive review of Lp(a) that also focuses on clinical studies examining the relationship between Lp(a) levels and CV risk.
Akaike M, Azuma H, Kagawa A, et al. Effect of aspirin treatment on serum concentrations of lipoprotein(a) in patients with atherosclerotic diseases. Clin Chem. 2002;48(9):1454–9.
Creider JC, Hegele RA, Joy TR. Niacin: another look at an underutilized lipid-lowering medication. Nat Rev Endocrinol. 2012;8(9):517–28.
The Coronary Drug Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA. 1975;231(4):360–81.
Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245–55.
Capuzzi DM, Guyton JR, Morgan JM, et al. Efficacy and safety of an extended-release niacin (Niaspan): a long-term study. Am J Cardiol. 1998;82(12A):74U–81. discussion 85U-86U.
Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J Intern Med. 1989;226(4):271–6.
Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575–9.
Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–12.
Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol. 2013;61(4):440–6.
Thompson GR, Barbir M, Davies D, et al. Efficacy criteria and cholesterol targets for LDL apheresis. Atherosclerosis. 2010;208(2):317–21.
McGowan MP. Emerging low-density lipoprotein (LDL) therapies: management of severely elevated LDL cholesterol—the role of LDL-apheresis. J Clin Lipidol. 2013;7(3 Suppl):S21–6.
Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice—evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145–284.
Leebmann J, Roeseler E, Julius U, et al. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567–76.
Thompson GR. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198(2):247–55.
Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med. 2009;6(3):229–39.
Taskinen MR, Puolakka J, Pyorala T, et al. Hormone replacement therapy lowers plasma Lp(a) concentrations. Comparison of cyclic transdermal and continuous estrogen-progestin regimens. Arterioscler Thromb Vasc Biol. 1996;16(10):1215–21.
Espeland MA, Marcovina SM, Miller V, et al. Effect of postmenopausal hormone therapy on lipoprotein(a) concentration. PEPI Investigators. Postmenopausal Estrogen/Progestin Interventions. Circulation. 1998;97(10):979–86.
Soma MR, Meschia M, Bruschi F, et al. Hormonal agents used in lowering lipoprotein(a). Chem Phys Lipids. 1994;67–68:345–50.
Sacks FM, McPherson R, Walsh BW. Effect of postmenopausal estrogen replacement on plasma Lp(a) lipoprotein concentrations. Arch Intern Med. 1994;154(10):1106–10.
Hoover-Plow J, Huang M. Lipoprotein(a) metabolism: potential sites for therapeutic targets. Metabolism. 2013;62(4):479–91.
Manson JE, Hsia J, Johnson KC, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349(6):523–34.
Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280(7):605–13.
Grady D, Herrington D, Bittner V, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA. 2002;288(1):49–57.
Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124–31.
KynamroTM (mipomersen sodium) injection [package insert]. Cambridge, MA; Genzyme. Revised January 2015.
Visser ME, Witztum JL, Stroes ES, Kastelein JJ. Antisense oligonucleotides for the treatment of dyslipidaemia. Eur Heart J. 2012;33(12):1451–8. This review summarizes major findings from phase 1-3 studies examining the effects of mipomersen, an anti-sense oligonucleotide to apoB, including effects on Lp(a).
Li N, Li Q, Tian XQ, Qian HY, Yang YJ. Mipomersen is a promising therapy in the management of hypercholesterolemia: a meta-analysis of randomized controlled trials. Am J Cardiovasc Drugs. 2014;14(5):367–76.
Juxtapid (lomitapide) capsules [package insert]. Cambridge, MA; Aegerion Pharmaceuticals. Revised August, 2014.
Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5(8):497–505.
Goldberg AC. Emerging low-density lipoprotein therapies: microsomal triglyceride transfer protein inhibitors. J Clin Lipidol. 2013;7(3 Suppl):S16–20.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–6.
Urban D, Poss J, Bohm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62(16):1401–8. This paper examines the therapeutic implications of PCSK9 inhibition on CV risk reduction.
Sniderman AD, Tsimikas S, Fazio S. The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol. 2014;63(19):1935–47.
Stein EA, Swergold GD. Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr Atheroscler Rep. 2013;15(3):310.
Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63(13):1278–88.
Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114(5):711–5.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–15.
REVEAL: randomized evaluation of the effects of anacetrapib through lipid-modification. https://clinicaltrials.gov/ct2/show/NCT01252953.
Mohammadpour AH, Akhlaghi F. Future of cholesteryl ester transfer protein (CETP) inhibitors: a pharmacological perspective. Clin Pharmacokinet. 2013;52(8):615–26.
ACCELERATE (Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition With Evacetrapib in Patients at a High-Risk for Vascular Outcomes). https://clinicaltrials.gov/ct2/show/study/NCT01687998?term=NCT01687998&rank=1 (ClinicalTrials.gov Identifier: NCT01687998).
Angelin B, Rudling M. Lipid lowering with thyroid hormone and thyromimetics. Curr Opin Lipidol. 2010;21(6):499–506.
Ladenson PW, Kristensen JD, Ridgway EC, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362(10):906–16.
Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109(7):961–71.
Chennamsetty I, Claudel T, Kostner KM, et al. Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest. 2011;121(9):3724–34.
Maeda S, Abe A, Seishima M, Makino K, Noma A, Kawade M. Transient changes of serum lipoprotein(a) as an acute phase protein. Atherosclerosis. 1989;78(2–3):145–50.
Berthold HK, Laudes M, Krone W, Gouni-Berthold I. Association between the interleukin-6 promoter polymorphism -174G/C and serum lipoprotein(a) concentrations in humans. PLoS One. 2011;6(9):e24719.
Schultz O, Oberhauser F, Saech J, et al. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS One. 2010;5(12):e14328.
Merki E, Graham M, Taleb A, et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57(15):1611–21.
Compliance with Ethics Guidelines
Conflict of Interest
Danielle Duffy received grants from Amgen, Forest Laboratories, Regeneron Pharmaceuticals, Aegerion Pharmaceuticals and Roche/Genentech; personal fees from AstraZeneca and Genzyme; and serves on the Board of Directors for Northeast Lipid Association and Southeastern Pennsylvania Chapter of the American Heart Association.
Lillian C. Man and Erik Kelly declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Nonstatin Drugs
Rights and permissions
About this article

Cite this article
Man, L.C., Kelly, E. & Duffy, D. Targeting Lipoprotein (a): an Evolving Therapeutic Landscape. Curr Atheroscler Rep 17, 25 (2015). https://doi.org/10.1007/s11883-015-0502-0
Published:
DOI: https://doi.org/10.1007/s11883-015-0502-0