
Abstract
Purpose of Review
Allergy and asthma are growing problems in the developed world. The accelerated increase of these diseases may be related to microbiome modification that leads to aberrant activation of Toll-like receptors (TLRs). Current research supports the concept that changes in microbial communities in early life impact TLR activation, resulting in an altered risk for the development of asthma and allergies.
Recent Findings
Prenatal and early childhood events that generate microbiome modification are closely related with TLR activation. Early childhood exposure to a rich array of TLR agonists, particularly lipopolysaccharide, strongly predicts protection against allergic disease later in life even when other lifestyle factors are accounted for. Genetic deletion of TLR signaling components in mice results in reduced function of tolerogenic cell populations in the gut. In contrast, weak TLR signaling can promote allergic sensitization later in life.
Summary
This review summarizes the role of TLR signaling in microbiome-mediated protection against allergy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
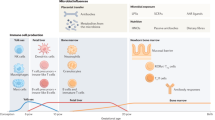
Allergic disease is the sixth leading cause of chronic illness in the USA. [1]. The unprecedented rise of allergy and asthma in the developed world, which has occurred in just a couple of generations, cannot be explained by genetic changes in the population over this short time frame [2]. Allergy has been described as a maladaptive immune response to innocuous environmental antigens, such as pollen, peanut, and animal dander [3]. The most likely culprits in explaining the rapid and recent rise of allergic disease incidence are environmental influences that include a loss of microbiota diversity. For example, there is evidence that indicates that gut microbiome modulates the activities of helper T cell subsets (Th1 and Th2) that can impact the development of immune tolerance [4]. Environmental effects such as exposure to pollution, living in environments with low endotoxin (urban environment), or smoking (Fig. 1) can collectively lead to immune activation and epigenetic modification [2, 3].

TLR activation. TLRs 1, 2, 4, 5, and 6 have been implicated in the development of allergies. Different ligands have been reported to activate a diverse set of TLRs. The principal signaling pathway that TLRs use is through MyD88-TRAF6 cascade that leads to NF-κB activation to regulate the transcription of inflammatory genes. TLRs, Toll-like receptors; MyD88, myeloid differentiation primary response gene 88; TRAF6, TNF receptor associated factor 6; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
The molecular basis of the allergic process has not been entirely established. However, several mechanisms have been proposed, among them are disruption in function and structure of epithelial barriers. In normal conditions, dendritic cells (DC) can sample antigens through the intact epithelial barrier by dendritic cell extension across epithelial cells and tight junction proteins [5], while in a damaged epithelial barrier, antigens can penetrate the underlying epithelium and activate the innate and adaptive immune response [5, 6]. Another proposed mechanism is that allergens activate protease receptors on epithelial cells, thereby inducing innate cytokines that drive T helper 2-like immunity [6]. The maturation and activation of innate cells are driven by TLR signaling for correct APC activation and to initiate the proper immune environment [7]. In allergic individuals, APCs capture antigen, migrate to the draining lymph node, and polarize T cells to become reactive against environmental allergens. Antigen-specific T cells infiltrate the tissue and produce cytokines (Th2) that promote classic symptoms of allergy [8]. Polarized Th2 T cells support B cell class switching to IgE, and infiltration of mast cells and eosinophils [5, 6]. Initiation of the classic Th2 immune response produces a positive reinforcement cycle of IL-4, IL-5, and IL-13 expression by T cells and innate lymphoid cells (ILCs). The production of innate cytokines IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) from the surrounding epithelial cells can both initiate and perpetuate the ongoing Th2-type responses [9]. The maturation and activation of the innate immune cells are driven in part by TLRs that are necessary to maintain immune homeostasis, but can also promote allergic responses when activated in the improper environment [7].
TLRs are a family of transmembrane protein receptors essential for the appropriate activation of the innate immune system, are evolutionarily preserved, and contain binding domains of leucine-rich repeat motifs. TLRs are one of the first lines of defense and activated by pathogen-associated molecular patterns (PAMPs), which are structurally conserved molecules that occur in many pathogens but not the host. Eleven TLRs have been identified in humans (TLR1 to TLR11), and all are functional except for TLR11 [7, 8]. TLRs are differentially located on the cell surface or in endosomes on both immune and non-immune cells critical to initiate the adaptive immune response, including epithelial cells, neutrophils, natural killer cells, and APC. Following TLR activation by their ligands, APCs increase their expression of costimulatory molecules (MHC, CD80, CD86, and CD40) and cytokines such as IL-6 are produced that can intervene in the suppressor activity of the Tregs [10]. Activated dendritic cells can also produce cytokines like IL-12 that help activate the clonal expansion of T cells [11]. TLRs also have an essential role in the maturation of dendritic cells [12] that participate in the development of healthy immune tolerance later in life. As TLR ligands are found both on beneficial and pathogenic microbes, the final result of TLR activation is dependent on cell type, location, and signal strength.
This review will cover how interactions between different TLRs, and their respective ligands, can either protect against or enhance the allergic process depending on location and stage of life (Fig. 1). It is now well documented that the gut microbiome plays an essential role in the development of the immune response, and it is becoming increasingly clear that both maternal influences and the early life microbiome that develops in the neonate have crucial roles in the development of allergies. An important component of microbiome-mediated protection appears to include TLR stimulation necessary to properly activate and mature regulatory APCs and T cells to avoid development of allergy.
Prenatal and Early Childhood Microbiome-TLR Interactions and the Susceptibility to Asthma and Allergy
Exposure to factors that affect the susceptibility of an individual to an allergic disease begin before birth, starting with the environment and allergy status of the mother. Microbes have been described in the placenta, amniotic fluid, fetal membrane, umbilical cord blood, and meconium [13,14,15,16]. The maternal-fetal interface is an active immune site, where the mother must maintain fetal tolerance but still protect against infection. Natural killer cells, dendritic cells, and macrophages infiltrate the endometrium and trophoblast regions and become activated during fetal development [17, 18]. The complex interactions between microbes, the maternal immune system, and developing fetus can have far-reaching consequences on childhood sensitivity to development of atopic disease.
Prenatal exposure to an environment rich in microbial compounds (including endotoxin), such as is found in rural and farm communities, reduces the prevalence of childhood allergy [19, 20•, 21]. Children of mothers with allergic disease who were supplemented with Lactobacillus rhamnosus and Bifidobacterium longum during pregnancy and the first 2 months of breastfeeding demonstrated decreased incidence of eczema and skin prick sensitivity to allergens at the age of 2 compared to mothers given placebo [22]. A decreased allergy incidence was found in similar studies, where prenatal probiotic supplementation was associated with microbial alteration detected in infant meconium compared to placebo control [22, 23]. These studies imply that a healthy microbiome during pregnancy can impact the development of allergy in the offspring. While the mechanism of probiotic-mediated protection is incompletely understood, studies in mice suggest that maternal TLR signaling is required to convey protective effects to the offspring. Conrad and colleagues demonstrated that maternal exposure to Acinetobacter lwoffi F78 protected the murine offspring against an allergic airway disease model, but protection was lost entirely if the challenged mothers were TLR2, TLR3, TLR4, TLR7, and TLR9 deficient (−/−) [24••]. Even though the offspring carried one functional allele of the TLRs from the father, they were not protected, demonstrating that TLR signaling in the mother was required for a protective effect. These studies suggest that the interaction between microbial products and the maternal immune system is essential to induce protective effects to the offspring.
Regulation of immune responses early in life appears to be central to controlling the development of allergic diseases. Since Tregs are involved in the early stage of the immune programming and their suppressive activity is needed to maintain a balance of the immune response, understanding the regulation of their development is essential to understanding the allergic disease. It has been described that the TLR2 and TLR4 pathways upregulate Treg development and activation [25] and that TLRs are expressed in both human and murine Tregs, suggesting direct TLR signaling for modulating Tregs function [26]. Previous studies using human cord blood mononuclear cells (HCBMC), which are reported to contain functional Tregs [26, 27], showed that cells obtained from infants of allergic mothers had decreased numbers of Tregs, and lower expression of Treg-associated genes accompanied by decreased suppressive function of the Tregs [28•]. HCBMC obtained from infants of allergic mothers had decreased production of IL-10 and IFN-γ, but increased production of IL-13, when stimulated by TLR2 ligand peptidoglycans (PPG) [28•]. Thus, maternal allergy could impair Treg development and function via TLR2 and TLR4 in the infants and increase susceptibility to allergic diseases [28•]. These latter findings were confirmed in studies using HCBMC from neonates stimulated with TLR2 or TLR4 ligands that found infants who developed eczema had a decreased percentage of Tregs and IL-10 cytokine production compared to HCBMC samples from non-allergic infants [27]. The altered cytokine response was accompanied by a lower percentage of Tregs in the samples from infants with atopic sensitization after, but not before, TLR2 stimulation. It was proposed that deficient Treg responses to microbial stimuli at the time of birth might contribute to increased risk of allergic diseases in the first year of life [27]. Thus, it appears that early activation of the immune system via TLR contributes to the development of Treg cells that can modify the immune environment.
Early Establishment of a Rich Microbiome Protects Against Allergic Disease
The microbiome composition and its diversity increase within the first years of life and determine the characteristics of the adult microbiome [29]. Since microbial components can provide strong TLR activating signals, they provide the earliest environmental stimuli to shape the immune responses. Microbial communities can be influenced by the use of pharmaceuticals, nutrients, microbial exposure, and infection [29, 30]. In a study that used human data from multiple countries, the prevalence of atopic sensitization and asthma was significantly lower in children who grew up on farms compared to those who did not with a stronger protective effect observed if mothers were also active on the farm. Interestingly, farm children had higher leukocyte gene expression of TLR2, TLR4, and CD14 (multifunctional receptor for endotoxin and other bacterial wall components) [21]. The importance of environmental microbial exposure was exemplified in a recent study, which compared allergy and asthma incidence in two closely related communities, the Amish and Hutterites. Despite sharing many similarities such as genetic ancestry, diet, family size, and long intervals of breastfeeding, Amish communities had much lower rates of childhood asthma and allergy compared to Huttertie communities. Protection in the Amish communities was closely associated with farming and increased levels of household endotoxin, diverse microbiome, and enhanced innate immune signaling; in contrast, Hutterite households had low endotoxin and reduced microbiome due to the purposefully separation of the community from the farm [31••]. The innate immune signals that were associated with protection are the same signals driven by TLR activation. Thus, understanding how the microbiome and their products (TLR ligands) influence the development of appropriate responses in susceptible populations will be critical.
The hypothesis unifying these findings is that increased microbiome diversity prepares the immune system for appropriate responses later in life by the recognition of commensal bacteria via TLRs and conditions the immune response to develop a balanced, rather than allergic, immune response. One of the most studied pathways linking the microbiome and immune regulation is microbial lipopolysaccharide (LPS) that activates TLR4. Peripheral blood mononuclear cells (PBMC) collected from children exposed to high levels of LPS in early life had decreased production of IL-10, IFN-γ, IL-12, and TNF-α when stimulated with LPS compared to children exposed to low levels of endotoxin. These results suggest that environmental exposure has a significant role in the development of tolerance to environmental stimuli [32]. It also has been suggested that the difference in TLR4 activation could be due to differences in LPS isoforms; the main difference between isoforms has been described in the TLR4-binding lipid A, which is the primary inducer of immunological response to LPS; and difference between variants is in the extent of acylation of the lipid A [33]. The penta-acylated lipid A isoform confers a degree of TLR-4 inhibition, while the hexa- or hepta-acylated lipid A variants are considered a potent stimulator of TLR4 signaling that leads to a Th1 response [33, 34].
Innate immune development is further impacted by early exposure to environmental triggers that alter molecular mechanisms of TLR-mediated cytokine production. For example, upon an infant’s initial exposure to LPS shortly after birth, intestinal epithelial cells become hypo-responsive to subsequent TLR stimulation, presumably to facilitate microbial colonization and host-microbe homeostasis [35]. Together, these studies suggest that increased microbial exposure prevents the development of allergic diseases through TLR regulation, and this effect is stronger if the exposure begins at prenatal stages and continues during the first years of life.
Recognition of Gut Microbes by TLR Supports Treg Expansion
Depletion of the microbiota with antibiotics sensitizes mice to allergy, whereas reconstitution of germ-free mice with protective microbiota protects against allergy [36, 37•, 38]. The beneficial effects of healthy microbiome are complex and diverse but include the production of beneficial metabolites that dampen systemic inflammation, including short-chain fatty acids and polyunsaturated fatty acids [39, 40]. Many of the anti-inflammatory effects of diverse gut microbiome are attenuated in mice with genetic deletions in TLR signaling components, suggesting that TLR-mediated recognition of commensal bacteria is essential for some of the protective effects of the microbiome [38, 41, 42, 43•]. For example, an influential study found that TLR4 and TLR5 ligation induced A proliferation-inducing ligand (APRIL) production in intestinal epithelial cells, which supported B cell class switching from IgM to IgA2 in the intestine, an essential component of tolerance against mucosal antigens [44]. Other groups have reported reduced tolerogenic mechanisms in mice with a genetic deletion in TLR signaling components, such as decreased production of IL-10 in lung interstitial macrophages of MyD88−/− and TLR4−/− mice [45], and reduced numbers of CD25+ regulatory T cells in the blood and spleen of MyD88−/− mice [46]. Current evidence points to a model where host-microbiome interactions through TLR recognition is critical for expanding regulatory subsets which protect against allergy, both in the gut and at distal sites such as the airway and skin [47]. However, the mechanism of how, or which components of the microbiome activate the TLR molecules, is unknown. Thus, a strategy to appropriately activate particular TLRs early in life may provide a protective response to generate a tolerogenic environment.
DC require microbial ligand activation via TLR signaling in order to generate regulatory T cells in the gut and the periphery. Transgenic mice engineered to have dendritic cell-specific TRAF6 deficiency (a primary component of TLR signaling) unexpectedly developed spontaneous Th2 inflammation and fibrosis in the gut that was associated with fewer intestinal Tregs in vivo. In vitro, TRAF6-deficient DC induced low levels of IL-2 and had a reduced capacity to induce Foxp3 expression in CD4 T cells [42••]. CD103+ DC play a critical role in homeostasis by capturing allergens, migrating to lymph nodes, and inducing tolerance. Intestinal CD103+ DC from TLR4−/− mice showed a reduced capacity to migrate to lymph nodes following oral antigen challenge and had a reduced ability to convert Tregs in vitro [48••]. These findings are complementary to other studies that show CD103+ DC sample the luminal contents of the gut in a TLR-dependent manner [49]. Thus, gut DC have a reduced capacity to promote tolerance to the antigen if they cannot recognize gut bacteria through TLRs.
Several groups have reported that T cell intrinsic TLR signaling plays a role in suppressing inflammation [35]. Transgenic mice with Foxp3-specific MyD88 deletion had reduced Treg and T follicular helper populations in the intestine and reduced total gut IgA and showed that TLR1/2-STAT3 signaling was necessary for optimal Treg cells [43•]. Two additional papers reported that T cell intrinsic TLR2 signaling actively promotes the production of IL-10 and the expression of CD25 and FoxP3 on T cells, and implicated both microbial (lipoproteins and peptidoglycan) and endogenous (human 60-kDa heat shock protein) ligands [50, 51]. These murine studies have been corroborated by in vitro analysis of T cells collected from asthmatic humans, where it was reported that increased expression of TLR2 and TLR4 on the surface of Tregs was associated with the enhanced suppressive ability [52]. These mechanistic studies suggest that early exposure to microbial ligands is crucial for developing regulatory immune cell populations toward potential allergens encountered later in life. Individuals who did not develop these protective populations are vulnerable to sensitization against innocuous antigens.
TLR and Food Allergy
In the past two decades, there has been an unprecedented rise in children that develop a food allergy. Currently, this statistic is at 1 in 13 children. The increased incidence of food allergy closely mirrors the expansion of other atopic diseases including allergy, eczema, and wheeze, suggesting a shared mechanism. The appropriate immune response is for gut DC and macrophages to induce strong tolerance to soluble antigens found in the diet by activating Tregs. It has long been known that antigens delivered orally induce tolerance at secondary challenge sites such as the lung and skin. Food allergy, therefore, represents a severe disruption of normal tolerogenic mechanisms [53••]. It has been proposed that sensitization against food allergens may occur at a different site, such as the skin. Like other allergy models, healthy gut flora protects against food allergy in mice through a complex and multifactorial mechanism [38]. Gnotobiotic studies in mice have demonstrated that colonization of the gut with protective strains of bacteria, notably from the Clostridia class, protects against food allergy, whereas depletion of bacteria with antibiotics sensitizes mice to food allergy [54].
Recent work supports an active role in TLR signaling in the microbiome-dependent protection against food allergy. Oral supplementation of pregnant mice with the probiotic Lactobacillus rhamnosus was found to protect pups against an OVA food allergy model and was associated with increased IgA+ cells in the intestine [55]. Pups from probiotic-supplemented mothers also had increased TLR2 expression in the small intestine, suggesting a protective role in TLR2-mediated immune activation. Consistent with this, a study reported that TLR4 deficiency increases the susceptibility to food allergy in numerous strains of mice, although the mechanism of susceptibility was unclear [56]. More recent work suggests that Treg maturation is central to the protective effects of the microbiome and TLR signaling. Wild-type mice that were orally gavaged with the skin contact sensitizer dinitrofluorobenzene (DNFB) were protected from a subsequent topical challenge, but protection was reduced in both germ-free and TLR4-deficient mice [48••]. Furthermore, MyD88−/− and TLR2−/− mice have reduced capacity to imprint gut-homing of Tregs, a critical step in developing intestinal tolerance [54]. Together, these studies suggest that TLR-APC interactions are critical for inducing tolerance to oral antigens.
Weak TLR Signaling Promotes Allergic Asthma
While early life exposure to microbial ligands is protective against allergy later in life, the same ligands play a critical role in sensitization against antigens encountered in the airway [57]. For example, early childhood exposure to endotoxin protects against allergy, but home endotoxin levels can be positively correlated with asthma in individuals over 18 years of age [58]. Low doses of endotoxin are sufficient to sensitize against allergen in allergic airway disease and a significant component of the house dust extract (HDE) model [6]. Interestingly, TLR4 signaling on both dendritic cells and radio-resistant structural cells contributes cumulatively to HDE allergy [59••, 60]. Furthermore, protease from Aspergillus oryzae can generate endogenous TLR4 ligands from fibrinogen cleavage products and can promote allergy by stimulating TLR4 allowing allergic sensitization [61•, 62•]. These studies further suggest that exposure to environmental irritants might promote asthma exacerbations through activation of TLRs.
Analysis of lung microbiome composition garners further evidence that TLR signaling plays a pathogenic role in airway hyperreactivity. The lungs of healthy individuals are colonized with bacteria [63•]. In healthy individuals, Provetella is the primary strain which colonizes the lung. In individuals with asthma, there is an outgrowth of Haemophilus, Streptococcus, and Moraxella subspecies [64]. The development of infantile wheezing in neonates delivered vaginally was lower compared to cesarean section (CS) [65] and was associated with decreased production of TNF-α and IL-6 in cord blood mononuclear cells in response to TLR1/2 stimulation in CS infants. Differential microbial communities were observed in the airways of the infants born by CS compared with natural delivery, with increased detection of Streptococcus pneumoniae, Staphylococcus aureus, and Moraxella catarrhalis. A correlation between CS delivery and increased risk of wheezing was observed at 1 year of age, suggesting that CS delivery increased the risk of wheeze by decreasing perinatal infant cytokine responses to TLR1/2 stimulation [65]. The critical role of microbiome diversity during the first month of life in the prevention of allergies has been reported [66], and the decreased microbial exposure that CS generated has been linked with the abnormal development of immunity [67]. Interestingly, in vitro studies demonstrated that asthma-associated pathogens Haemophilus spp. and Moraxella spp. elicit higher cytokine production from human DC and THP-1-derived macrophages than commensal lung bacteria [68]. The LPS structure of Haemophilus and Moraxella elicits 10-fold higher TLR4 signaling compared to commensal bacteria. Bacterial species associated with healthy lung microbiome elicit minimal TLR responses, whereas bacterial species associated with asthma and COPD can elicit more robust TLR responses. Mice experimentally inoculated with the asthma-associated proteobacteria exhibited enhanced neutrophilia and cytokine production compared to mice inoculated with commensal Provetella [69]. Provetella bacteria promoted leukocyte recruitment to the lungs in a TLR2-dependent manner, but this recruitment was nonpathogenic. The enhanced pro-inflammatory capacity of pathobionts and the inert immunological properties of healthy commensals suggest that minimal TLR signaling in the airway can protect against allergy.
Strong TLR Signaling Protects Against Allergic Airway Disease
While trace levels of TLR ligands in the airway promote airway sensitization and Th2-type immunity, pharmacological doses of the same ligands can protect against allergy [27, 70, 71]. The differential outcome that TLR ligands generate in allergic sensitization has created apparent contradictions in the literature, where some authors report that TLR ligands are pathogenic while others report they are protective. In a seminal study, low-dose LPS exacerbated allergy, whereas high-dose LPS protected against eosinophilia and production of Th2 cytokines in association with enhanced interferon gamma production [71]. Subsequent studies demonstrated that repeated administration of low-dose LPS has a protective effect by suppression of cytokine production in epithelial cells [72•]. While low-dose flagellin (TLR5 ligand) enhanced OVA-induced allergy in mice, a high dose of flagellin protected mice against allergy by generating regulatory dendritic cells and T cells dependent on CD25+ cells Treg cells [73••]. The differential effects of flagellin in allergic airway disease can also be observed in other studies [74] and showed that high dose is associated with accumulation of regulatory cell populations [75]. Probiotics also protect against allergic airway disease using intranasal administration of Lactococcus lactis G121 to protect mice in an OVA asthma model [76] through TLR3 and TLR8 activation resulting in IL-10 and IL-12 production [77•]. The distinct response to TLR stimulation has historically been described as a shift from Th2- to Th1-type immunity as the strength of stimulation is increased. More recent work suggests a model where Th2 responses are dampened not only by a competing Th1 response, but by the activation of Tregs, and IL-10-producing DC and macrophages [47].
The use of strong TLR ligands to treat allergic asthma has also been examined in pre-clinical models and in clinical trials. In particular, unmethylated CpG was shown to have a particularly strong effect in blocking the onset and maintenance of allergic responses as well as reversing established disease in experimental allergic asthma [78, 79]. However, clinical trials using TLR9 stimulatory CpG compounds have been variable and mixed with the overall phase 2 studies showing little efficacy or benefit in moderate to severe asthmatics [80,81,82,83]. Additional studies using other endosomal TLRs, TLR3, and TLR7 agonists have also shown promising effects in vivo in animal models [84,85,86, 87]. Thus, there continues to be significant interest in using individual or combinations of TLR agonists to provide strong activation to modulate asthmatic/allergic diseases.
Conclusions
It appears that the maternal and early childhood microbiome can generate long-lasting alterations in the TLR responses that can lead to either a decreased or increased predisposition to asthma and allergies. The loss of microbiota diversity may be a significant cause of the rise of allergy, atopy, and asthma in industrialized countries. Mechanistic studies in mice demonstrate that immune cell subsets that protect against allergy fail to fully develop if protective microbiota or the microbe-TLR signaling pathways are compromised. Individuals who lack early, rich microbiota exposure and the associated protective immune subsets become vulnerable to sensitization against innocuous antigens in the environment. Thus, the timing, intensity, and type of stimuli are likely to have a significant impact on the development of protective vs. pathogenic responses. The same antigens and allergens that promote tolerance in early life can become pathogenic later in life in vulnerable individuals (Fig. 2).

Proposed relationship between TLR activation, age, and susceptibility to allergy. Individuals who are exposed to a rich variety of TLR agonists, such as living on a farm or growing up with pets, during early childhood are driven toward a tolerogenic immune response that protects against allergy later in life. Peripheral Tregs, mucosal IgA production, and IL-10 production appear to regulate allergic disease development. In contrast, individuals with low exposure to microbial products lack these protective regulatory subsets and are vulnerable to allergic disease. Later in life, diverse TLR signaling from the environment can shift this pro-inflammatory state of the immune system into pathogenic allergic disease
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major Importance
CDC. Allergies. 2017. http://www.cdc.gov/healthcommunication/toolstemplates/entertainmented/tips/Allergies.html.
Begin P, Nadeau KC. Epigenetic regulation of asthma and allergic disease. Allergy Asthma Clin Immunol. 2014;10(1):27. https://doi.org/10.1186/1710-1492-10-27.
Palmer LJ, Burton PR, Faux JA, James AL, Musk AW, Cookson WO. Independent inheritance of serum immunoglobulin E concentrations and airway responsiveness. Am J Respir Crit Care Med. 2000;161(6):1836–43. https://doi.org/10.1164/ajrccm.161.6.9805104.
Riiser A. The human microbiome, asthma, and allergy. Allergy Asthma Clin Immunol. 2015;11:35. https://doi.org/10.1186/s13223-015-0102-0.
van Ree R, Hummelshoj L, Plantinga M, Poulsen LK, Swindle E. Allergic sensitization: host-immune factors. Clin Transl Allergy. 2014;4(1):12. https://doi.org/10.1186/2045-7022-4-12.
Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410–6. https://doi.org/10.1038/nm.1946.
Gangloff SC, Guenounou M. Toll-like receptors and immune response in allergic disease. Clin Rev Allergy Immunol. 2004;26(2):115–25. https://doi.org/10.1007/s12016-004-0006-0.
Takeda K, Akira S. Roles of Toll-like receptors in innate immune responses. Genes Cells. 2001;6(9):733–42.
Lau MY, Dharmage SC, Burgess JA, Win AK, Lowe AJ, Lodge C, et al. The interaction between farming/rural environment and TLR2, TLR4, TLR6 and CD14 genetic polymorphisms in relation to early- and late-onset asthma. Sci Rep. 2017;7:43681. https://doi.org/10.1038/srep43681.
Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033–6. https://doi.org/10.1126/science.1078231.
Mitchell TC, Hildeman D, Kedl RM, Teague TK, Schaefer BC, White J, et al. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat Immunol. 2001;2(5):397–402. https://doi.org/10.1038/87692.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80. https://doi.org/10.1038/90609.
Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65. https://doi.org/10.1126/scitranslmed.3008599.
Prince AL, Ma J, Kannan PS, Alvarez M, Gisslen T, Harris RA, et al. The placental membrane microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am J Obstet Gynecol. 2016;214(5):627 e1–e16. https://doi.org/10.1016/j.ajog.2016.01.193.
Chu DM, Antony KM, Ma J, Prince AL, Showalter L, Moller M, et al. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016;8(1):77. https://doi.org/10.1186/s13073-016-0330-z.
Fonseca W, Lukacs NW, Ptaschinski C. Factors affecting the immunity to respiratory syncytial virus: from epigenetics to microbiome. Front Immunol. 2018;9:226. https://doi.org/10.3389/fimmu.2018.00226.
Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342(20):1500–7. https://doi.org/10.1056/NEJM200005183422007.
Koga K, Mor G. Toll-like receptors at the maternal-fetal interface in normal pregnancy and pregnancy disorders. Am J Reprod Immunol. 2010;63(6):587–600. https://doi.org/10.1111/j.1600-0897.2010.00848.x.
Karvonen AM, Hyvarinen A, Gehring U, Korppi M, Doekes G, Riedler J, et al. Exposure to microbial agents in house dust and wheezing, atopic dermatitis and atopic sensitization in early childhood: a birth cohort study in rural areas. Clin Exp Allergy. 2012;42(8):1246–56. https://doi.org/10.1111/j.1365-2222.2012.04002.x.
• Karvonen AM, Hyvarinen A, Rintala H, Korppi M, Taubel M, Doekes G, et al. Quantity and diversity of environmental microbial exposure and development of asthma: a birth cohort study. Allergy. 2014;69(8):1092–101. https://doi.org/10.1111/all.12439 This study demostrated the importance of early life microbial exposure in the development of asthma.
Ege MJ, Bieli C, Frei R, van Strien RT, Riedler J, Ublagger E, et al. Prenatal farm exposure is related to the expression of receptors of the innate immunity and to atopic sensitization in school-age children. J Allergy Clin Immunol. 2006;117(4):817–23. https://doi.org/10.1016/j.jaci.2005.12.1307.
Rautava S, Kainonen E, Salminen S, Isolauri E. Maternal probiotic supplementation during pregnancy and breast-feeding reduces the risk of eczema in the infant. J Allergy Clin Immunol. 2012;130(6):1355–60. https://doi.org/10.1016/j.jaci.2012.09.003.
Rautava S, Luoto R, Salminen S, Isolauri E. Microbial contact during pregnancy, intestinal colonization and human disease. Nat Rev Gastroenterol Hepatol. 2012;9(10):565–76. https://doi.org/10.1038/nrgastro.2012.144.
•• Conrad ML, Ferstl R, Teich R, Brand S, Blumer N, Yildirim AO, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med. 2009;206(13):2869–77. https://doi.org/10.1084/jem.20090845 This work highlights the importance of prenatal Toll-like receptors activation by microbial exposure and the protective effect on the development of allergies.
Lluis A, Depner M, Gaugler B, Saas P, Casaca VI, Raedler D, et al. Increased regulatory T-cell numbers are associated with farm milk exposure and lower atopic sensitization and asthma in childhood. J Allergy Clin Immunol. 2014;133(2):551–9. https://doi.org/10.1016/j.jaci.2013.06.034.
Dai J, Liu B, Li Z. Regulatory T cells and Toll-like receptors: what is the missing link? Int Immunopharmacol. 2009;9(5):528–33. https://doi.org/10.1016/j.intimp.2009.01.027.
Schaub B, Liu J, Hoppler S, Haug S, Sattler C, Lluis A, et al. Impairment of T-regulatory cells in cord blood of atopic mothers. J Allergy Clin Immunol. 2008;121(6):1491–9, 9 e1-13. https://doi.org/10.1016/j.jaci.2008.04.010.
• Meng SS, Gao R, Yan BD, Ren J, Wu F, Chen P, et al. Maternal allergic disease history affects childhood allergy development through impairment of neonatal regulatory T-cells. Respir Res. 2016;17(1):114. https://doi.org/10.1186/s12931-016-0430-8. This study found that a history of maternal allergy was associated with altered Treg phenotype in neonates, demonstrating that susceptibility to allergy can begin prenatally.
Rodriguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. 2015;26:26050. https://doi.org/10.3402/mehd.v26.26050.
Levin AM, Sitarik AR, Havstad SL, Fujimura KE, Wegienka G, Cassidy-Bushrow AE, et al. Joint effects of pregnancy, sociocultural, and environmental factors on early life gut microbiome structure and diversity. Sci Rep. 2016;6:31775. https://doi.org/10.1038/srep31775.
•• Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, et al. Innate immunity and asthma risk in Amish and Hutterite farm children. N Engl J Med. 2016;375(5):411–21. https://doi.org/10.1056/NEJMoa1508749 This study demonstrated the importance of microbial exposure and the development of allergies in two similar genetic communities.
Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347(12):869–77. https://doi.org/10.1056/NEJMoa020057.
Vuillermin PJ, Macia L, Nanan R, Tang ML, Collier F, Brix S. The maternal microbiome during pregnancy and allergic disease in the offspring. Semin Immunopathol. 2017;39(6):669–75. https://doi.org/10.1007/s00281-017-0652-y.
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–5. https://doi.org/10.1038/nature07830.
Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37(5):771–83. https://doi.org/10.1016/j.immuni.2012.10.014.
Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149(7):1578–93. https://doi.org/10.1016/j.cell.2012.04.037.
• Kozakova H, Schwarzer M, Tuckova L, Srutkova D, Czarnowska E, Rosiak I, et al. Colonization of germ-free mice with a mixture of three lactobacillus strains enhances the integrity of gut mucosa and ameliorates allergic sensitization. Cell Mol Immunol. 2016;13(2):251–62. https://doi.org/10.1038/cmi.2015.09 This study clearly demonstrates a cause-and-effect relationship between colonization with protective bacteria and protection against allergy.
Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, Mazmanian SK, et al. Commensal bacteria protect against food allergen sensitization. PNAS. 2014;111(36):13145–50. https://doi.org/10.1073/pnas.1412008111.
Fonseca W, Lucey K, Jang S, Fujimura KE, Rasky A, Ting HA, et al. Lactobacillus johnsonii supplementation attenuates respiratory viral infection via metabolic reprogramming and immune cell modulation. Mucosal Immunol. 2017;10(6):1569–80. https://doi.org/10.1038/mi.2017.13.
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–66. https://doi.org/10.1038/nm.3444.
Bashir MEH, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol. 2004;172(11):6978–87. https://doi.org/10.4049/jimmunol.172.11.6978.
Han D, Walsh Matthew C, Cejas Pedro J, Dang Nicholas N, Kim Youngmi F, Kim J, et al. Dendritic cell expression of the signaling molecule TRAF6 is critical for gut microbiota-dependent immune tolerance. Immunity. 2013;38(6):1211–22. https://doi.org/10.1016/j.immuni.2013.05.012.
• Wang S, Charbonnier L-M, Rivas MN, Georgiev P, Li N, Gerber G, et al. MyD88 adaptor-dependent microbial sensing by regulatory T cells promotes mucosal tolerance and enforces commensalism. Immunity. 2015;43(2):289–303. https://doi.org/10.1016/j.immuni.2015.06.014. This study demonstrated that T-cell intrinsic TLR signaling was required to maintain intestinal homeostasis and prevent inflammation. It provided evidence that a protective microbiome limits inflammation through direct interaction with T cells.
He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26(6):812–26. https://doi.org/10.1016/j.immuni.2007.04.014.
Kawano H, Kayama H, Nakama T, Hashimoto T, Umemoto E, Takeda K. IL-10-producing lung interstitial macrophages prevent neutrophilic asthma. Int Immunol. 2016;28(10):489–501. https://doi.org/10.1093/intimm/dxw012.
Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116(2):485–94. https://doi.org/10.1172/JCI25439.
Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol. 2017;18(10):1076–83. https://doi.org/10.1038/ni.3829.
•• Hacini-Rachinel F, Gomez de Agüero M, Kanjarawi R, Moro-Sibilot L, Le Luduec J-B, Macari C, et al. Intestinal dendritic cell licensing through Toll-like receptor 4 is required for oral tolerance in allergic contact dermatitis. J Allergy Clin Immunol. 2018;141(1):163–70. https://doi.org/10.1016/j.jaci.2017.02.022 This study demonstrates that dendritic cell-intrinsic TLR signaling is necessary to promote tolerance against oral antigens at peripheral sites. It provides mechanistic insights to the breakdown of oral tolerance.
Farache J, Koren I, Milo I, Gurevich I, Kim K-W, Zigmond E, et al. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity. 2013;38(3):581–95. https://doi.org/10.1016/j.immuni.2013.01.009.
Jun JC, Jones MB, Oswald DM, Sim ES, Jonnalagadda AR, Kreisman LSC, et al. T cell-intrinsic TLR2 stimulation promotes IL-10 expression and suppressive activity by CD45RbHi T cells. PLoS One. 2017;12(7):e0180688. https://doi.org/10.1371/journal.pone.0180688.
Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116(7):2022–32. https://doi.org/10.1172/JCI28423.
Pace E, Di Sano C, Ferraro M, Bruno A, Caputo V, Gallina S, et al. Budesonide increases TLR4 and TLR2 expression in Treg lymphocytes of allergic asthmatics. Pulm Pharmacol Ther. 2015;32:93–100. https://doi.org/10.1016/j.pupt.2015.02.003.
•• Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science. 2016;351(6275):858–63. https://doi.org/10.1126/science.aac5560 This study demonstrates that dietary antigens sustain a diverse Treg repertoire in mucosal sites.
Wang S, Villablanca EJ, De Calisto J, Gomes DC, Nguyen DD, Mizoguchi E, et al. MyD88-dependent TLR1/2 signals educate dendritic cells with gut-specific imprinting properties. J Immunol. 2011;187(1):141–50. https://doi.org/10.4049/jimmunol.1003740.
Saliganti V, Kapila R, Sharma R, Kapila S. Feeding probiotic Lactobacillus rhamnosus (MTCC 5897) fermented milk to suckling mothers alleviates ovalbumin-induced allergic sensitisation in mice offspring. Br J Nutr. 2015;114(8):1168–79. https://doi.org/10.1017/S000711451500286X.
Berin MC, Zheng Y, Domaradzki M, Li XM, Sampson HA. Role of TLR4 in allergic sensitization to food proteins in mice. Allergy. 2006;61(1):64–71. https://doi.org/10.1111/j.1398-9995.2006.01012.x.
Thorburn AN, Tseng H-Y, Donovan C, Hansbro NG, Jarnicki AG, Foster PS, et al. TLR2, TLR4 AND MyD88 mediate allergic airway disease (AAD) and Streptococcus pneumoniae-induced suppression of AAD. PLoS One. 2016;11(6):e0156402. https://doi.org/10.1371/journal.pone.0156402.
Kain HL, Osusky R. Pars plana lensectomy in pediatric cataract. Klin Monatsbl Augenheilkd. 1992;200(5):451–3. https://doi.org/10.1055/s-2008-1045790.
•• McAlees JW, Whitehead GS, Harley ITW, Cappelletti M, Rewerts CL, Holdcroft AM, et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 2015;8(4):863–73. https://doi.org/10.1038/mi.2014.117 The TLR4 compartment study demonstrates that the role of TLR4 signaling and airway sensitization is an additive effect. Numerous cells relay sensitization signals through TLR4 that culminate in an allergic response.
Thomas SY, Whitehead GS, Takaku M, Ward JM, Xu X, Nakano K, et al. MyD88-dependent dendritic and epithelial cell crosstalk orchestrates immune responses to allergens. Mucosal Immunol. 2017;11:796–810. https://doi.org/10.1038/mi.2017.84.
• Cho M, Lee J-E, Lim H, Shin H-W, Khalmuratova R, Choi G, et al. Fibrinogen cleavage products and Toll-like receptor 4 promote the generation of programmed cell death 1 ligand 2–positive dendritic cells in allergic asthma. J Allergy Clin Immunol. 2017;142:530–541.e6. https://doi.org/10.1016/j.jaci.2017.09.019. This study provides novel insight on how proteases can promote allergic sensitization. The authors show that proteases can act on endogenous proteins to produce TLR4 ligands.
• Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science. 2013;341(6147):792–6. https://doi.org/10.1126/science.1240342.
• Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 2017;10(2):299–306. https://doi.org/10.1038/mi.2016.108 This review discusses, in depth, the role of the respiratory tract microbiome. The authors of this manuscript were instrumental in showing that the respiratory tract is not sterile.
O'Connor GT, Lynch SV, Bloomberg GR, Kattan M, Wood RA, Gergen PJ, et al. Early-life home environment and risk of asthma among inner-city children. J Allergy Clin Immunol. 2018;141(4):1468–75. https://doi.org/10.1016/j.jaci.2017.06.040.
Liao SL, Tsai MH, Yao TC, Hua MC, Yeh KW, Chiu CY, et al. Caesarean section is associated with reduced perinatal cytokine response, increased risk of bacterial colonization in the airway, and infantile wheezing. Sci Rep. 2017;7(1):9053. https://doi.org/10.1038/s41598-017-07894-2.
Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016;22(10):1187–91. https://doi.org/10.1038/nm.4176.
Neu J, Rushing J. Cesarean versus vaginal delivery: long-term infant outcomes and the hygiene hypothesis. Clin Perinatol. 2011;38(2):321–31. https://doi.org/10.1016/j.clp.2011.03.008.
Bernasconi E, Pattaroni C, Koutsokera A, Pison C, Kessler R, Benden C, et al. Airway microbiota determines innate cell inflammatory or tissue remodeling profiles in lung transplantation. Am J Respir Crit Care Med. 2016;194(10):1252–63. https://doi.org/10.1164/rccm.201512-2424OC.
Larsen JM, Musavian HS, Butt TM, Ingvorsen C, Thysen AH, Brix S. Chronic obstructive pulmonary disease and asthma-associated Proteobacteria, but not commensal Prevotella spp., promote Toll-like receptor 2-independent lung inflammation and pathology. Immunology. 2015;144(2):333–42. https://doi.org/10.1111/imm.12376.
Remot A, Descamps D, Noordine M-L, Boukadiri A, Mathieu E, Robert V, et al. Bacteria isolated from lung modulate asthma susceptibility in mice. ISME J. 2017;11(5):1061–74. https://doi.org/10.1038/ismej.2016.181.
Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4–dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196(12):1645–51. https://doi.org/10.1084/jem.20021340.
• Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349(6252):1106–10. https://doi.org/10.1126/science.aac6623 The farm dust study shows a novel mechanism by which repeated airway exposure of endotoxin can protect against allergy. Earlier reports have focused on Th1/Th2 polarization and the role of adaptive immunity, rather than tissue-intrinsic effects.
•• Shim J-U, Lee SE, Hwang W, Lee C, Park J-W, Sohn J-H, et al. Flagellin suppresses experimental asthma by generating regulatory dendritic cells and T cells. J Allergy Clin Immunol. 2016;137(2):426–35. https://doi.org/10.1016/j.jaci.2015.07.010 This study clearly demonstrates the importance of dose in determining whether TLR5 signaling is protective, or pathogenic, in allergic airway sensitization.
Lee LM, Ji M, Sinha M, Dong MB, Ren X, Wang Y, et al. Determinants of divergent adaptive immune responses after airway sensitization with ligands of Toll-like receptor 5 or Toll-like receptor 9. PLoS One. 2016;11(12):e0167693. https://doi.org/10.1371/journal.pone.0167693.
Kitzmüller C, Kalser J, Mutschlechner S, Hauser M, Zlabinger GJ, Ferreira F, et al. Fusion proteins of flagellin and the major birch pollen allergen Bet v 1 show enhanced immunogenicity, reduced allergenicity, and intrinsic adjuvanticity. J Allergy Clin Immunol. 2018;141(1):293–9.e6. https://doi.org/10.1016/j.jaci.2017.02.044.
Debarry J, Garn H, Hanuszkiewicz A, Dickgreber N, Blümer N, Mutius EV, et al. Acinetobacter lwoffii and Lactococcus lactis strains isolated from farm cowsheds possess strong allergy-protective properties. J Allergy Clin Immunol. 2007;119(6):1514–21. https://doi.org/10.1016/j.jaci.2007.03.023.
• Stein K, Brand S, Jenckel A, Sigmund A, Chen ZJ, Kirschning CJ, et al. Endosomal recognition of Lactococcus lactis G121 and its RNA by dendritic cells is key to its allergy-protective effects. J Allergy Clin Immunol. 2017;139(2):667–78.e5. https://doi.org/10.1016/j.jaci.2016.06.018. This study demonstrated that maturation of tolerogenic dendritic cells is dependent on TLR signaling.
Sudo N, Aiba Y, Oyama N, Yu XN, Matsunaga M, Koga Y, et al. Dietary nucleic acid and intestinal microbiota synergistically promote a shift in the Th1/Th2 balance toward Th1-skewed immunity. Int Arch Allergy Immunol. 2004;135(2):132–5. https://doi.org/10.1159/000080655.
Carver JD, Pimentel B, Cox WI, Barness LA. Dietary nucleotide effects upon immune function in infants. Pediatrics. 1991;88(2):359–63.
Gauvreau GM, Hessel EM, Boulet LP, Coffman RL, O'Byrne PM. Immunostimulatory sequences regulate interferon-inducible genes but not allergic airway responses. Am J Respir Crit Care Med. 2006;174(1):15–20. https://doi.org/10.1164/rccm.200601-057OC.
Senti G, Johansen P, Haug S, Bull C, Gottschaller C, Muller P, et al. Use of A-type CpG oligodeoxynucleotides as an adjuvant in allergen-specific immunotherapy in humans: a phase I/IIa clinical trial. Clin Exp Allergy. 2009;39(4):562–70. https://doi.org/10.1111/j.1365-2222.2008.03191.x.
Beeh KM, Kanniess F, Wagner F, Schilder C, Naudts I, Hammann-Haenni A, et al. The novel TLR-9 agonist QbG10 shows clinical efficacy in persistent allergic asthma. J Allergy Clin Immunol. 2013;131(3):866–74. https://doi.org/10.1016/j.jaci.2012.12.1561.
Casale TB, Cole J, Beck E, Vogelmeier CF, Willers J, Lassen C, et al. CYT003, a TLR9 agonist, in persistent allergic asthma - a randomized placebo-controlled phase 2b study. Allergy. 2015;70(9):1160–8. https://doi.org/10.1111/all.12663.
Aryan Z, Rezaei N. Toll-like receptors as targets for allergen immunotherapy. Curr Opin Allergy Clin Immunol. 2015;15(6):568–74. https://doi.org/10.1097/ACI.0000000000000212.
Starkhammar M, Kumlien Georen S, Swedin L, Dahlen SE, Adner M, Cardell LO. Intranasal administration of poly(I:C) and LPS in BALB/c mice induces airway hyperresponsiveness and inflammation via different pathways. PLoS One. 2012;7(2):e32110. https://doi.org/10.1371/journal.pone.0032110.
Sel S, Wegmann M, Sel S, Bauer S, Garn H, Alber G, et al. Immunomodulatory effects of viral TLR ligands on experimental asthma depend on the additive effects of IL-12 and IL-10. J Immunol. 2007;178(12):7805–13.
Kaufman EH, Fryer AD, Jacoby DB. Toll-like receptor 7 agonists are potent and rapid bronchodilators in guinea pigs. J Allergy Clin Immunol. 2011;127(2):462–9. https://doi.org/10.1016/j.jaci.2010.10.029.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Basic and Applied Science
Rights and permissions
About this article

Cite this article
Michels, K.R., Lukacs, N.W. & Fonseca, W. TLR Activation and Allergic Disease: Early Life Microbiome and Treatment. Curr Allergy Asthma Rep 18, 61 (2018). https://doi.org/10.1007/s11882-018-0815-5
Published:
DOI: https://doi.org/10.1007/s11882-018-0815-5