
Abstract
Purpose of Review
Many genetic conditions predispose affected individuals to opportunistic infections. A number of immunodeficiency diseases, including genetic defects termed Mendelian susceptibility to mycobacterial disease (MSMD), permit infection from many different strains of mycobacteria that would otherwise not cause disease. These include tuberculous and nontuberculous mycobacteria, and bacille Calmette-Guérin vaccine (BCG). Patients may present with infections from other organisms that depend on macrophage function for containment. Defects in multiple genes in the IL-12 and NFKB signaling pathways can cause the MSMD phenotype, some of which include IL12RB1, IL12B, IKBKG, ISG15, IFNGR1, IFNGR2, CYBB, TYK2, IRF8, and STAT1.
Recent Findings
Multiple autosomal recessive and dominant, and 2 X-linked recessive gene defects resulting in the MSMD phenotype have been reported, and others await discovery. This review presents the known gene defects and describes clinical findings that result from the mutations.
Summary
If MSMD is suspected, a careful clinical history and examination and basic immunodeficiency screening tests will narrow the differential diagnosis. A specific diagnosis requires more sophisticated laboratory investigation. Genetic testing permits a definitive diagnosis, permitting genetic counseling. Mild cases respond well to appropriate antibiotic therapy, whereas severe disease may require hematopoietic stem cell transplantation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mycobacteria are found in soil and water. In humans, they are commensal organisms, the nontuberculous mycobacteria being epithelial colonists of the gastrointestinal, respiratory, and genitourinary systems, as well as the skin. Limbs of the innate and adaptive immune systems normally keep the organisms in check. Either hyporesponsiveness or hyperresponsiveness to these organisms can result in clinical disease [1]. In some countries, bacille Calmette-Guérin (BCG) vaccine, a live vaccine developed from Mycobacterium bovis, is used to prevent tuberculosis. Individuals with certain primary immunodeficiency diseases (including severe combined immunodeficiency, hyper IgM syndrome, and chronic granulomatous disease) can develop disseminated mycobacterial infection following immunization. An interesting series of inherited or sporadic defects that mostly affect the interferon-gamma (IFNγ) pathway have been termed Mendelian susceptibility to mycobacterial disease (MSMD). Nontuberculous mycobacterial disease can also occur in patients with secondary immunodeficiency, including HIV-1 disease, malignancy, immunosuppressive pharmacotherapy, or cystic fibrosis [2•].
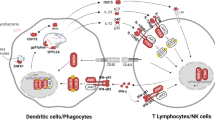
The professional phagocytes (neutrophils, dendritic cells, macrophages, monocytes) of the innate immune system are key players, whose pattern recognition receptors (PRRs) can interact with mycobacterial danger-associated molecular patterns (DAMPs). Mycobacterial DAMPs are found in the cell wall. PRRs important in mycobacterial recognition include Toll-like receptors (TLR-2, TLR-4, TLR-6, TLR-9), NOD2, MINCLE and MARCO, Dectin-1, Dectin-2, Dectin-3, DC-SIGN, Galectin-3, CD36, mannose-binding lectin (MBL), and NLRP3 [1]. Although there is some functional redundancy in the PRR system, loss of function defects, or absence of innate immune cells bearing them, can increase susceptibility to mycobacterial infection and, in some cases, also infection with Salmonella sp., Candida sp., viruses, and other organisms. In the adaptive immune system, cytotoxic (CD8+) and helper (CD4+) T cells provide host defense against mycobacterial infection. T cell deficiency, primary or acquired, permits infection. Some MSMD defects are found in the NFKB signaling pathway. The interleukin-12 (IL-12) signaling pathway, with production of interferon-gamma (IFNγ), is particularly important for host resistance to mycobacteria, and defects in this complex pathway account for most known causes of MSMD. Among other roles in the innate and adaptive immune system, IFNγ activates macrophages, enhancing their ability to kill intracellular organisms, such as mycobacteria and Salmonella sp. Other organisms handled by macrophages include Klebsiella sp., Histoplasma sp., Paracoccidioides sp., Coccidioides sp., and Cryptococcus sp. [3•].
Infection from weakly virulent Salmonella sp. and mycobacteria such as nontuberculous mycobacteria and BCG is uncommon to very rare. Some patients have mild disease, whereas others have life-threatening, difficult-to-treat, and sometimes fatal disease. Patients typically present with mycobacterial infections in childhood [4••]. Some adult patients presenting with the MSMD phenotype have autoantibodies to IFNγ [5•] or GATA2 deficiency. Here, we will concisely review the currently known genetic defects that may result in the MSMD phenotype (including some that are not usually classified as part of the MSMD family), discuss clinical features of the defects, following the classification and order given in Table 1, and comment on patient evaluation (Table 2) and therapy.
TLR-2
An important PRR of the innate immune system, Toll-like receptor 2 (TLR-2) recognizes mycobacterial lipoproteins and is responsible for inducing IL-12 production in macrophages. Mutations can increase susceptibility to tuberculosis [9] and leprosy (M. leprae, M. lepromatosis).
IL-12/23Rβ1
Interleukin 12 (IL-12) is produced by dendritic cells, macrophages, and neutrophils in response to activating signals. It promotes differentiation of naïve T cells into Th1 cells and stimulates T cells and NK cells to produce IFNγ and TNFα. Production of IFNγ enhances macrophage phagocytosis, which is important in the host defense against mycobacteria. The receptor is a heterodimer. The IL-12Rβ1 subunit binds with IL-12Rβ2 to form the IL-12 receptor, or with IL-23R to form the IL-23 receptor. A defect in the IL12RB1 gene (which produces IL-12/23Rβ1) causes immunodeficiency 30, the most common known cause of MSMD [3•]. Affected patients are susceptible to infection with bacille Calmette-Guérin (BCG), other atypical mycobacteria, M. tuberculosis, and Salmonella sp. [10•]. Some patients have presented with associated enteropathy and hypogammaglobulinemia [11]. The IL-12Rβ1 variation database lists 220 individuals and 192 unique DNA variants [12]. Inheritance is autosomal recessive.
IL-12/23 p40 Subunit
IL-12 is a heterodimer encoded by 2 separate genes, IL12A (p35 subunit) and IL12B (p40 subunit). The IL-12 p40 subunit also heterodimerizes with the IL-23 p19 subunit (IL23A) to form IL-23. Binding of IL-12 to its receptor activates helper T cells and NK cells. A defect in IL12B (IL-12 p40 subunit), causes immunodeficiency 29, another commonly reported cause of MSMD. Patients typically have BCG disease and/or salmonellosis, but tuberculosis and atypical mycobacterial disease have been noted. At least 49 patients have been reported [13•]. Inheritance is autosomal recessive.
IFNγR1
The receptor for human interferon-gamma (IFNγ), the type II interferon, is a heterodimer of IFNγR1 (that binds IFNγ) and IFNγR2. IFNγ activates macrophages, and plasma IFNγ levels can be elevated in patients with IFNγR1 or IFNγR2 deficiency [14]. IFNγR1 deficiency can present in autosomal recessive (AR) and autosomal dominant (AD) forms. AR deficiency (immunodeficiency 27A) presents with severe, early-onset infections with atypical mycobacteria or BCG. Tuberculosis and salmonellosis have been reported. Patients with the AD deficiency (immunodeficiency 27B) have less severe, later onset mycobacterial infections, but are more prone to Mycobacterium avium complex osteomyelitis [15, 16].
IFNγR2
Defects in IFNγR2 (immunodeficiency 28), which is involved in signal transduction, also result in severe, early-onset mycobacterial infections. In 1998, Dorman and Holland reported a child with disseminated M. fortuitum and M. avium complex infection and a mutation in the extracellular domain of IFNγR2 [17]. Other mutations have been reported [18].
STAT1
Signal from the IFNγR, as well as the type I interferon receptor, is transduced via a JAK/STAT pathway that uses JAK1 (Janus kinase 1) and JAK2, and STAT1 (signal transducer and activator of transcription 1). JAK phosphorylation of STAT1 produces a homodimer that binds to DNA sequences in the nuclear gamma interferon-activated site. STAT1 deficiency can present in autosomal dominant (immunodeficiency 31A) and autosomal recessive (immunodeficiency 31B) inheritance patterns. The AD form affects IFNγR signal transduction, producing relatively mild disease from BCG, M. avium complex, and TB. The AR form also affects signal transduction from the type I interferon receptor, producing severe mycobacterial and viral disease.
JAK1 (OMIM 147795)
Janus kinase 1 (JAK1) is a tyrosine kinase that adds a phosphate group to a tyrosine amino acid on substrate proteins. JAK1 is required for interferon type I (IFNα, IFNβ), and type II (IFNγ) signal transduction. A JAK1 deficiency resulted in atypical mycobacterial disease in a 22-year-old Pakistani male from consanguineous parents [19]. Inheritance is autosomal recessive.
ISG15
ISG15 is a ubiquitin-like modifier induced by interferon α and interferon β, resulting in conjugation to many cellular proteins, and release from monocytes to cause NK cell activation and cytotoxicity [20]. In the absence of ISG15, the amount of IFNγ production is reduced, which may account for the increased susceptibility to mycobacterial disease, as found in immunodeficiency 38. Bogunovic et al. reported 3 patients who developed severe BCG and mycobacterial disease [21]. Intracranial calcification is also reported [22]. Inheritance is autosomal recessive.
RORC
RAR-related orphan receptor C (RORC) encodes for the transcription factor RORγ. The RORγt isoform promotes differentiation of thymocytes into Th17 cells. Homozygous mutations of RORC were discovered in 3 patients from different consanguineous families. RORC deficiency causes immunodeficiency 42, which presents in infancy with candida and mycobacterial infections. Patients have impaired production of IL-17A, IL-17F, and IL-22, and an apparent defect in IFNγ production in response to BCG and IL-12 [23]. Inheritance is autosomal recessive.
GATA2 (“MonoMAC Syndrome”)
SLC11A1/NRAMP1
Solute carrier family 11, member 1 (SLC11A1), also known as natural resistance-associated macrophage protein 1 (NRAMP1), is a macrophage membrane protein that also appears to be involved in neutrophil function. Deficiency has been reported to confer susceptibility to tuberculosis and leprosy, as well as rheumatoid arthritis [28].
IRF8
Interferon regulatory factor 8 (IRF8) is a member of a family of transcription factors that regulate type I interferon gene expression [29]. It is only present in immune cells [30]. Deficiency can present as autosomal dominant immunodeficiency 32A, as found in two individuals of Italian descent with disseminated BCG disease and a loss of a dendritic cell population. An autosomal recessive form, termed immunodeficiency 32B, causes monocyte and dendritic cell deficiency with resulting severe infections, including disseminated BCG and candidiasis [31].
ΤΥΚ2
Tyrosine kinase 2 (TYK2) stabilizes cell surface expression of the interferon α and β receptor subunit 1 (IFNAR1) [32]. TYK2 deficiency produces immunodeficiency 35, reported in a 22-year-old Japanese male with hyper IgE syndrome diagnosed at age 22 months with a BCG infection [33]. He had a history of infections with viruses, fungi, and mycobacteria, and Salmonella sp. gastroenteritis at age 15. His healthy, consanguineous parents were heterozygous for the mutation. A few other patients have been reported, and elevated IgE is not a consistent finding. Inheritance is autosomal recessive.
NFKBIA/IκBα
Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha (NFKBIA, or IκBα), inhibits the NFKB complex (along with NFKBIB), inactivating the transcription factor NF-κB (NFKB) by preventing nuclear translocation [34]. NFKBIA gain of function mutations decreases the production of cytokines and interferons, producing anhidrotic ectodermal dysplasia and susceptibility to various kinds of infections, including a few recently reported patients with BCG and mycobacterial disease [35,36,37]. Inheritance is autosomal dominant (chromosome 14).
IKBKB/IKKβ
Inhibitor of kappa light chain gene enhancer in B cells, kinase of, beta (IKBKB/IKKβ/IKK2) and IKBKA/IKKα/IKK1, is a subunit of IκB kinase (IKK), which phosphorylates IκB proteins and permits activation of NF-κB (NFKB). IKBKB and IKBKA associate with IKBKG (NEMO) to form IKK. Mutations in IKBKB cause immunodeficiency 15, which presents in early life with severe viral, fungal, and bacterial infections, as well as MSMD. Immunoglobulin levels are low, although T and B cells are present. Burns et al. presented a female, born to consanguineous parents, diagnosed with a generalized rash with disseminated BCG at age 3 months. PCR was positive for M. tuberculosis complex. Exome sequencing revealed an IKBKB gene mutation [38]. Other mutations have been reported [39].
IKBKG/IKKγ/NEMO
Inhibitor of kappa light chain gene enhancer in B cells, kinase of, gamma (IKBKG, IKKγ), also termed NF-κB essential modulator (NEMO), is a subunit of IκB kinase (IKK). Mutations in NEMO can present with varied features, including immunodeficiency 33, X-linked recessive susceptibility to mycobacterial disease. This is probably due to defective IL-12 production, causing low levels of IFNγ. Patients typically present with M. avium complex disease, and some have dental abnormalities, including conical teeth. [40, 41].
CARMIL2/RLTPR
RGD-, leucine-rich repeat-, tropomodulin domain-, and proline-rich domain-containing protein (RLTPR), also known as capping protein regulator and myosin 1 linker 2 (CARMIL2), is vital for T cell CD28 co-stimulation in T cells, acting in a scaffolding role to bridge CD28 to CARD11 and the NFKB pathway. Six patients with CARMIL2 gene mutations were reported by Wang et al.; two of which developed mycobacterial infections. At age 8, 1 patient was diagnosed with multifocal tuberculosis within the cervical lymph nodes, respiratory tract, and digestive tract. The other patient developed miliary tuberculosis at 9 years old. Mucocutaneous candidiasis was also reported [42].
CYBB
Neutrophils kill ingested organisms by using NADPH-oxidase, which assembled from several subunits. One subunit, cytochrome b(-245) or p91-phox, is a heterodimer of a beta subunit (CYBB) and an alpha subunit (CYBA). Deficiency of either subunit can cause chronic granulomatosis disease (CGD), but CYBB deficiency is not uniformly associated with a defective neutrophil oxidative burst assay. A CYBB gene defect causes immunodeficiency 34 without CGD features as described by Bustamante et al., who presented a group of 7 male patients with CYBB missense mutations leading to recurrent BCG disease or recurrent tuberculosis [43]. Inheritance is x-linked recessive.
Conclusions
Infection with BCG or nontuberculous mycobacteria in a child is a rare event that should prompt a diligent search for an underlying immunodeficiency, particularly if there have also been infections with Salmonella sp. or Candida sp. The list of known causes of MSMD (Table 1) is daunting, and other gene defects await discovery, so evaluating a patient with recurrent mycobacterial infections is a challenging task [6••]. The differential diagnosis can be narrowed somewhat by a detailed history and examination, as well as some basic laboratory tests (Table 2). Because many defects are inherited, a detailed family history (including careful questioning to determine consanguinuity) is warranted. Routine laboratory screening tests, such as a CBC with differential, immunoglobulin levels (G, A, M, E), lymphocyte cytofluorometry (T, B, NK), and HIV1 testing, warrant consideration. Lymphocyte functional testing may be indicated.
An absence or marked reduction in monocytes numbers on routine CBC with differential suggests GATA2 deficiency (MonoMAC). Elevation of plasma IFNγ suggests a deficiency in a component of the IFNγ receptor heterodimer [14]. Some forms of specialized testing, including genetic testing panels, are available from commercial reference laboratories, but detailed testing is most effectively coordinated by a primary immunodeficiency center with expert immunology and genetics laboratories [6••]. In some cases, some form of DNA sequencing (such as whole genome sequencing or whole exome sequencing) will be indicated, since new gene defects await discovery. Genetic confirmation of the diagnosis is essential, not only for accurate diagnosis but also for effective genetic counseling. In countries that offer BCG immunization, genetic testing of family members of MSMD patients would be warranted prior to immunization.
Some of the milder forms of MSMD require no treatment other than appropriate antibiotics. Recombinant interferon-gamma injections have also been used in patients with defects that affect IFNγ production, although the product does not have FDA approval for this indication. The more severe forms have been treated with hematopoietic stem cell transplantation. Gene therapy and other approaches are on the horizon [44•].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major Importance
Robinson RT, Huppler AR. The Goldilocks model of immune symbiosis with Mycobacteria and Candida colonizers. Cytokine. 2017;97:49–65. https://doi.org/10.1016/j.cyto.2017.05.015.
• Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. 2015;264(1):103–20. https://doi.org/10.1111/imr.12272. This review, not limited to MSMD, focuses on predisposing factors for childhood tuberculosis.
• Louvain de Souza T, de Souza Campos Fernandes RC, Azevedo da Silva J, Gomes Alves Junior V, Gomes Coelho A, Souza Faria AC, et al. Microbial disease spectrum linked to a novel IL-12Rbeta1 N-terminal signal peptide stop-gain homozygous mutation with paradoxical receptor cell-surface expression. Front Microbiol. 2017;8:616. https://doi.org/10.3389/fmicb.2017.00616. This case report describes the clinical presentation of 4 children with MSMD and reviews the features of IL12RB1 inactivation.
•• Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26(6):454–70. https://doi.org/10.1016/j.smim.2014.09.008. This is a very detailed review of the causes of MSMD.
• Lee WI, Huang JL, Wu TS, Lee MH, Chen IJ, Yu KH, et al. Patients with inhibitory and neutralizing auto-antibodies to interferon-gamma resemble the sporadic adult-onset phenotype of Mendelian susceptibility to mycobacterial disease (MSMD) lacking Bacille Calmette-Guerin (BCG)-induced diseases. Immunobiology. 2013;218(5):762–71. https://doi.org/10.1016/j.imbio.2012.08.281. Autoantibodies to IFNγ can produce the clinical phenotype of MSMD in adults.
•• Esteve-Sole A, Sologuren I, Martinez-Saavedra MT, Deya-Martinez A, Oleaga-Quintas C, Martinez-Barricarte R, et al. Laboratory evaluation of the IFN-gamma circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci. 2018;55(3):184–204. https://doi.org/10.1080/10408363.2018.1444580. This article reviews the basic science of the IFNγ circuit in the context of MSMD, and offers detailed advice for laboratory investigation of patients with suspected MSMD.
Online Mendelian Inheritance in Man, OMIM® [database on the Internet]. McKusick-Nathans Institute of Genetic Medicine. Available from: https://omim.org. Accessed: 1/3/18.
• Picard C, Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2017;38:96–128. https://doi.org/10.1007/s10875-017-0464-9. This is the most recent official list of primary immunodeficiency diseases recognized by the International Union of Immunological Societies. The list, which is frequently updated, has descriptions of the MSMD conditions.
Ogus AC, Yoldas B, Ozdemir T, Uguz A, Olcen S, Keser I, et al. The arg753-to-gln polymorphism of the human Toll-like receptor 2 gene in tuberculosis disease. Europ Resp J. 2004;23:219–23.
• de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine. 2010;89(6):381–402. https://doi.org/10.1097/MD.0b013e3181fdd832. This somewhat older article is a very large case series describing clinical features of IL12RB1 deficiency.
Arias AA, Perez-Velez CM, Orrego JC, Moncada-Velez M, Rojas JL, Wilches A, et al. Severe enteropathy and hypogammaglobulinemia complicating refractory mycobacterium tuberculosis complex disseminated disease in a child with IL-12Rbeta1 deficiency. J Clin Immunol. 2017;37(7):732–8. https://doi.org/10.1007/s10875-017-0435-1.
IL-12Rβ1 deficiency: mutation update and description of the IL12RB1 variation database [database on the Internet]. LOVD. 2013. Available from: https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php?select_db=IL12RB1. Accessed: April 2018.
• Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, et al. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine (Baltimore). 2013;92(2):109–22. https://doi.org/10.1097/MD.0b013e31828a01f9. Although IL12B (p40) deficiency is rare, this case series describes features of 49 patients with that disorder.
Fieschi C, Dupuis S, Picard C, Smith CIE, Holland SM, Casanova J-L. High levels of interferon gamma in the plasma of children with complete interferon gamma receptor deficiency. Pediatrics. 2001;107(4):E48.
Janssen R, Van Wengen A, Verhard E, De Boer T, Zomerdijk T, Ottenhoff TH, et al. Divergent role for TNF-alpha in IFN-gamma-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-gamma receptor 1 deficiency. J Immunol. 2002;169(7):3900–7.
Storgaard M, Varming K, Herlin T, Obel N. Novel mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infections. Scand J Immunol. 2006;64(2):137–9. https://doi.org/10.1111/j.1365-3083.2006.01775.x.
Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101(11):2364–9. https://doi.org/10.1172/JCI2901.
Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8):1729–37. https://doi.org/10.1084/jem.20071987.
Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun. 2016;7:13992. https://doi.org/10.1038/ncomms13992.
Meraro D, Gleit-Kielmanowicz M, Hauser H, Levi BZ. IFN-stimulated gene 15 is synergistically activated through interactions between the myelocyte/lymphocyte-specific transcription factors, PU.1, IFN regulatory factor-8/IFN consensus sequence binding protein, and IFN regulatory factor-4: characterization of a new subtype of IFN-stimulated response element. J Immunol. 2002;168(12):6224–31. https://doi.org/10.4049/jimmunol.168.12.6224.
Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012;337(6102):1684–8. https://doi.org/10.1126/science.1224026.
Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517:89–93.
Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349(6248):606–13. https://doi.org/10.1126/science.aaa4282.
Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, Olivier KN, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519–29. https://doi.org/10.1182/blood-2009-03-208629.
Calvo KR, Vinh DC, Maric I, Wang W, Noel P, Stetler-Stevenson M, et al. Myelodysplasia in autosomal dominant and sporadic monocytopenia immunodeficiency syndrome: diagnostic features and clinical implications. Haematologica. 2011;96(8):1221–5. https://doi.org/10.3324/haematol.2011.041152.
Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–5. https://doi.org/10.1182/blood-2011-05-356352.
Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–8. https://doi.org/10.1182/blood-2011-06-360313.
Shaw M-A, Clayton D, Atkinson SE, Williams H, Miller N, Sibthorpe D, et al. Linkage of rheumatoid arthritis to the candidate gene NRAMP1 on 2q35. J Med Genet. 1996;33:672–7.
Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87(2):307–17.
Driggers PH, Elenbaas BA, An JB, Lee IJ, Ozato K. Two upstream elements activate transcription of a major histocompatibility complex class I gene in vitro. Nucleic Acids Res. 1992;20(10):2533–40.
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365(2):127–38. https://doi.org/10.1056/NEJMoa1100066.
Ragimbeau J, Dondi E, Alcover A, Eid P, Uze G, Pellegrini S. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 2003;22(3):537–47. https://doi.org/10.1093/emboj/cdg038.
Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745–55. https://doi.org/10.1016/j.immuni.2006.09.009.
Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95(6):749–58.
Staples E, Morillo-Gutierrez B, Davies J, Petersheim D, Massaad M, Slatter M, et al. Disseminated Mycobacterium malmoense and Salmonella infections associated with a novel variant in NFKBIA. J Clin Immunol. 2017;37(5):415–8. https://doi.org/10.1007/s10875-017-0390-x.
Lee AJ, Moncada-Vélez M, Picard C, Llanora G, Huang C, Abel L, et al. Severe mycobacterial diseases in a patient with GOF IκBα mutation without EDA. J Clin Immunol. 2016;36:12–5.
Yoshioka T, Nishikomori R, Hara J, Okada K, Hashii Y, Okafuji I, et al. Autosomal dominant anhidrotic ectodermal dysplasia with immunodeficiency caused by a novel NFKBIA mutation, p.Ser36Tyr, presents with mild ectodermal dysplasia and non-infectious systemic inflammation. J Clin Immunol. 2013;33(7):1165–74. https://doi.org/10.1007/s10875-013-9924-z.
Burns SO, Plagnol V, Gutierrez BM, Al Zahrani D, Curtis J, Gaspar M, et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKbeta. J Allergy Clin Immunol. 2014;134(1):215–8. https://doi.org/10.1016/j.jaci.2013.12.1093.
Nielsen C, Jakobsen MA, Larsen MJ, Muller AC, Hansen S, Lillevang ST, et al. Immunodeficiency associated with a nonsense mutation of IKBKB. J Clin Immunol. 2014;34(8):916–21. https://doi.org/10.1007/s10875-014-0097-1.
Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet. 2000;67(6):1555–62. https://doi.org/10.1086/316914.
Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203(7):1745–59.
Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016;213(11):2413–35. https://doi.org/10.1084/jem.20160576.
Bustamante J, Arias A, Vogt G, Picard C, Galicia L, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12(3):213–21.
• Haverkamp MH, van de Vosse E, van Dissel JT. Nontuberculous mycobacterial infections in children with inborn errors of the immune system. J Inf Secur. 2014;68(Suppl 1):S134–50. https://doi.org/10.1016/j.jinf.2013.09.024. An illustrated review of the basic science of IL-12, IFNγ, and IFNα signaling pathways with a detailed discussion of individual defects causing MSMD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Reed and Dolen declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pediatric Allergy and Immunology
Rights and permissions
About this article

Cite this article
Reed, B., Dolen, W.K. The Child with Recurrent Mycobacterial Disease. Curr Allergy Asthma Rep 18, 44 (2018). https://doi.org/10.1007/s11882-018-0797-3
Published:
DOI: https://doi.org/10.1007/s11882-018-0797-3